RELATO DE CASO
Síndrome de von Hippel-Lindau em um Serviço Privado de Câncer em São Paulo: Relato de Caso
Von Hippel-Lindau Syndrome in a Private Cancer Service in São Paulo: Case Report
Síndrome de von Hippel-Lindau en un Servicio Privado de Cáncer en São Paulo: Informe de Caso
doi: https://doi.org/10.32635/2176-9745.RBC.2023v69n1.2686
Letícia Marchioro Leandro da Costa¹; Ana Paula Bedin²; Nicolli Romualdo Coutinho³; Rafael Araujo Ferro do Lago4; Thaís Neves Macruz Oliveira5; Victor André de Almeida Zia6
1,3Centro Universitário de Volta Redonda (UniFOA). Volta Redonda (RJ), Brasil. E-mails: leticiamarchioro17@gmail.com; nicolli.rcoutinho@gmail.com. Orcid iD: https://orcid.org/0000-0002-1771-8372; Orcid iD: https://orcid.org/0000-0002-4375-721x
2,4Universidade Municipal de São Caetano do Sul. São Paulo (SP), Brasil. E-mails: ana-paula-bedin@hotmail.com; rafaellago_15@hotmail.com. Orcid iD: https://orcid.org/0000-0003-4464-6236; Orcid iD: https://orcid.org/0000-0003-3793-2744
5Universidade Nove de Julho. São Paulo (SP), Brasil. E-mail: thais.macruz@icloud.com. Orcid iD: https://orcid.org/0000-0002-0870-8121
6Universidade de São Paulo (USP). Ribeirão Preto (SP), Brasil. E-mail: victor.z@uol.com.br. Orcid iD: https://orcid.org/0000-0003-3558-9604
Endereço para correspondência: Letícia Marchioro Leandro da Costa. Rua Carlos Gomes, 85, Apto. 801 – Centro. Volta Redonda (RJ), Brasil. CEP 27250-620. E-mail: leticiamarchioro17@gmail.com
Introdução: A síndrome de von Hippel-Lindau (VHL) é uma patologia hereditária autossômica dominante que envolve o crescimento de tumores em diversas regiões do corpo humano em razão da mutação no gene VHL. Relato do caso: Paciente, sexo masculino, 38 anos, há três anos queixava-se de cefaleia recorrente, com piora progressiva. Foi diagnosticado com uma lesão em cerebelo cuja ressonância magnética cerebral encontrou uma formação expansiva na porção posteroinferior do hemisfério cerebelar esquerdo. Foi realizada tomografia multislice de abdome, que evidenciou formação nodular esplênica com realce marginal. A imagem da coluna cervical demonstrou pequeno nódulo localizado na medula cervical (intramedular) adjacente à vértebra cervical 3 (C3). Diante dos achados, o paciente foi submetido à ressecção macroscópica total da lesão do cerebelo, com laudo anatomopatológico de hemangioblastoma cerebelar grau 1, de acordo com a classificação da Organização Mundial da Saúde (OMS), que é um tumor benigno com baixa agressividade e recorrência. O teste imuno-histoquímico mostrou cluster of differentiation 34 (CD 34) positivo, índice de proliferação celular (Ki67) positivo (<5%), alfa inibina positiva e epithelial membrane antigen (EMA) negativo. Como o paciente não tinha história familiar de câncer, em função dos achados radiológicos, foi realizado sequenciamento de nova geração identificando a variante patogênica VHL c.292T>C, constatado em linhagem germinativa que, apesar do desconhecimento de história familiar positiva para a síndrome, confirmou o diagnóstico do paciente. Conclusão: O conjunto de achados clínicos e a variante no gene VHL confirmam o diagnóstico da síndrome.
Palavras-chave: doença de von Hippel-Lindau; mutação em linhagem germinativa; hemangioblastoma.
ABSTRACT
Introduction: Von Hippel-Lindau (VHL) syndrome is an autosomal dominant hereditary pathology that involves the growth of tumors in different regions of the human body due to mutation of the VHL gene. Case report: Male patient, 38 years old, complained of recurrent headache for 3 years, with progressive worsening. A lesion in the cerebellum was diagnosed, whose magnetic resonance imaging found an expansive formation in the posteroinferior portion of the left cerebellar hemisphere. Multislice tomography of the abdomen was performed, showing splenic nodular formation with marginal enhancement. Cervical spine imaging demonstrated a small nodule located in the cervical (intramedullary) cord adjacent to cervical vertebra 3 (C3). In view of the findings, the patient underwent total macroscopic resection of the cerebellar lesion, with an anatomopathological report of World Health Organization (WHO) grade 1 cerebellar hemangioblastoma, which is a benign tumor with lower risk of aggressiveness and recurrence. Immunohistochemical test showed positive cluster of differentiation 34 (CD34), cell proliferation index positive (Ki67) (<5%), positive alpha inhibin and epithelial membrane antigen (EMA) negative. As the patient had no family history of cancer, a new generation sequencing was performed due to the radiological findings, which identified the pathogenic variant VHL c.292T>C found in germ lineage; although the family was unaware of any past family history of the syndrome, the patient’s diagnosis was confirmed. Conclusion: The set of clinical findings and the variant in the VHL gene confirm the diagnosis of the syndrome.
Key words: von Hippel-Lindau disease; germ-line mutation; hemangioblastoma.
RESUMEN
Introducción: El síndrome de Von Hippel-Lindau (VHL) es una patología hereditaria autosómica dominante que consiste en el crecimiento de tumores en diferentes regiones del cuerpo humano debido a una mutación en el gen VHL. Informe del caso: Paciente, masculino, 38 años, consulta por cefalea recurrente desde hace 3 años, con empeoramiento progresivo. Se diagnosticó lesión en cerebelo, cuya resonancia magnética encontró una formación expansiva en la porción posteroinferior del hemisferio cerebeloso izquierdo. Se realizó tomografía multicorte de abdomen, que mostró formación nodular esplénica con realce marginal. Las imágenes de la columna cervical demostraron un pequeño nódulo ubicado en el cordón cervical (intramedular) adyacente a vértebra cervical 3 (C3). Ante los hallazgos se procedió a la resección macroscópica total de la lesión cerebelosa, con informe anatomopatológico de hemangioblastoma cerebeloso grado 1, de acuerdo con la clasificación de la Organización Mundial de Salud (OMS) que es un tumor benigno con baja agresividad y recurrencia. La prueba inmunohistoquímica mostró cluster of differentiation 34 (CD34) positivo, índice de proliferación celular (Ki67) positivo (<5%), alfa inhibina positivo y epithelial membrane antigen (EMA) negativo. Como el paciente no tenía antecedentes familiares de cáncer, debido a los hallazgos radiológicos, se realizó una secuenciación de nueva generación identificando la variante patogénica VHL c.292T>C, encontrada en el linaje germinal, que, a pesar del desconocimiento de antecedentes familiares positivos para el síndrome, confirmó el diagnóstico del paciente. Conclusión: El conjunto de hallazgos clínicos y la variante en el gen VHL confirman el diagnóstico del síndrome.
Palabras clave: enfermedad de von Hippel-Lindau; mutación de línea germinal; hemangioblastoma.
INTRODUÇÃO
A síndrome de von Hippel-Lindau (VHL) é uma patologia hereditária autossômica dominante que envolve o crescimento de tumores em diversas regiões do corpo humano1. A mutação ocorre no gene VHL, supressor tumoral, que está no lócus cromossômico 3p25.32,3. A doença gera uma predisposição para o desenvolvimento de neoplasias hipervascularizadas, sendo as mais comuns: hemangioblastomas de sistema nervoso central (SNC) e retina, carcinoma de células renais, cistos renais, feocromocitoma e tumores císticos e sólidos de pâncreas4.
Em 1991, Neumann classificou essa síndrome de acordo com a frequência de tumores5. Existem cinco subtipos descritos atualmente: o tipo 1, causado por deleções ou mutações truncadas, é caracterizado por apresentar alto risco para o carcinoma renal de células claras e hemangioblastoma de SNC e retina, e baixo risco para feocromocitoma, sendo a forma clínica mais comum; o tipo 1B, causado por exclusões genéticas contíguas que abrangem o VHL, apresenta alto risco para hemangioblastoma de SNC e retina e baixo risco para feocromocitoma e carcinoma renal de células claras; o tipo 2, causado por mutações missense, apresenta alto risco para o feocromocitoma, sendo dividido em: 2A – alto risco para feocromocitoma, hemangioblastoma de SNC e retina e baixo risco para carcinoma renal de células claras; 2B – alto risco para feocromocitoma, hemangioblastoma de SNC e retina e carcinoma de células claras; 2C – alto risco para feocromocitoma e baixo risco para hemangioblastoma de SNC e retina, e carcinoma renal de células claras6.
A expressividade da doença varia segundo o tipo de mutação encontrada. O carcinoma de células renais tem como subtipo mais comum o de células claras, e é uma causa frequente de morte7.
O diagnóstico clínico pode ser feito de duas maneiras: paciente com histórico familiar positivo e apenas uma manifestação clínica, hemangioblastoma em retina ou cerebelar por exemplo, e em pacientes sem histórico familiar, com duas ou mais manifestações clínicas características da síndrome7.
O objetivo do seguinte relato é esclarecer à comunidade médica sobre como a síndrome de VHL se apresenta na prática clínica, assim como facilitar seu reconhecimento e diagnóstico pelos profissionais.
RELATO DO CASO
O presente caso é de um paciente do sexo masculino, de 38 anos, previamente saudável. Há três anos da admissão, queixava-se de cefaleia recorrente, com piora progressiva. Por isso, procurou um neurologista, tendo sido diagnosticado com lesão em cerebelo e encaminhado para investigação complementar.
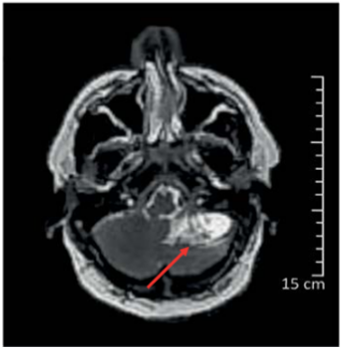
Uma ressonância magnética do crânio mostrou formação expansiva na porção posteroinferior do hemisfério cerebelar esquerdo de 27 × 26 × 25 mm e era caracterizada por isossinal em T1, hipersinal em T2/FLAIR, com aspecto multilobulado, e proeminente realce heterogêneo ao meio de contraste, caracterizando-se vascularização intralesional, conforme demonstrado na Figura 1.
|
|
|
Figura 1. Ressonância magnética de crânio demonstrando sítio de hemangioblastoma no hemisfério cerebelar esquerdo |
No abdome superior, foi realizada uma tomografia multislice que mostrou baço (cuja topografia, dimensões e densidade estavam normais) com formação nodular levemente lobulada e com realce marginal com até 2,9 cm. Os demais órgãos encontravam-se sem alterações significativas.
Foi realizada ainda uma ressonância magnética da coluna cervical, da coluna torácica e da coluna lombar que apresentou pequeno nódulo isointenso em T1 e hiperintenso em T2, com limites bem definidos, com realce ávido pelo meio de contraste, medindo 0,9 cm, localizado na medula cervical (intramedular) adjacente à vértebra cervical 3 (C3).
Diante dos achados, o paciente foi submetido a uma ressecção macroscópica total da lesão em cerebelo, com laudo de hemangioblastoma cerebelar grau 1 da Organização Mundial da Saúde (OMS)8, sendo um tumor benigno com menor risco de agressividade e recidiva. O teste imuno-histoquímico mostrou cluster os differentiation 34 (CD 34) positivo, índice de proliferação celular (Ki67) positivo (<5%), alfa inibina positiva e epithelial membrane antigen (EMA) negativo.
Em razão dos achados sugestivos para a síndrome de VHL, o paciente foi avaliado por geneticista que, diante da suspeita, solicitou sequenciamento de nova geração do gene VHL, no qual foi identificada a variante chr3:10142139 T>C (VHL c.292T>C, NM_000551), em heterozigose no gene VHL que promove a substituição do aminoácido tirosina no códon 98 por histidina (VHL p.Tyr98His)9. De acordo com os critérios da American College of Medical Genetics (ACGM)10, a variante chr3:10142139 T>C (VHL c.292T>C, NM_000551) é considerada patogênica.
Foi realizada outra ressonância magnética cranioencefálica de controle, que mostrou os sinais de craniotomia occipital bilateral, orifício de trepanação occipital à direita, redução volumétrica do hemisfério cerebelar esquerdo associado à proeminência das folias cerebelares, além de imagens com hipersinal em T2, predomínio de hipersinal com alguns focos de hipersinal em FLAIR de permeio, caracterizando áreas de gliose e focos de encefalomalácia.
Atualmente, o paciente mantém acompanhamento oftalmológico, neurológico (anual, baseado na avaliação clínica) e radiológico (bianual – ressonância de neuroeixo e abdome, podendo a ressonância ser substituída por tomografia de abdome ou ultrassom, quando a ressonância não estiver disponível), de acordo com os últimos guidelines11.
Este estudo atendeu à Resolução n.º 466/201212 do Conselho Nacional de Saúde e foi aprovado pelo Comitê de Ética em Pesquisa da Notre Dame Intermédica Saúde S.A. pela Invitare Pesquisa Clínica Auditoria e Consultoria Ltda, sob o número de parecer 4.869.492 (CAAE: 49966321.0.0000.8098).
DISCUSSÃO
A síndrome de VHL é uma patologia com herança autossômica dominante e defeitos no gene VHL localizado no braço curto do cromossomo 3. Latif et al.13 identificaram rearranjos no gene VHL, sendo uma parte por causa das deleções no gene, uma destas in-frame de três nucleotídeos. Crossey et al.14 identificaram 40 tipos diferentes de mutações, sendo as duas mais frequentes: arg238-to-gln e arg238-to-trp. Lenglet et al.15 identificaram mutações complexas em heterozigose no E1-prime do gene VHL, obtendo como resultado as substituições leu128-to-val (L128V) e leu138-reportsto-pro (L138P). O gene VHL codifica a proteína VHL (pVHL), supressora de tumor envolvida nas vias de sinalização celular. Existem duas isoformas de proteína VHL: VHL30 e VHL19 e ambas são importantes para os efeitos supressores de tumor4.
Majoritariamente, o indivíduo afetado herda o alelo mutado de genitor também portador de VHL (80% dos casos), os demais casos provêm de mutações novas16. A produção anormal da pVHL gera a transcrição de vários genes e o aumento de produção de fatores de crescimento, incluindo eritropoietina, fator de crescimento endotelial vascular, fator de crescimento derivado de plaquetas B e outros genes envolvidos na captação e metabolismo da glicose, levando à formação de cistos e tumores hipervasculares característicos de VHL17.
Trata-se de uma doença rara que representa um desafio para o diagnóstico médico em razão do aparecimento de tumores infrequentes que, ao serem observados de maneira isolada, acabam não levantando a suspeita da síndrome. Afeta 1:36.000 a 1:45.000 nascidos vivos e há prevalência de 1:38.0000 a 1:91.000. A maioria dos pacientes manifesta a síndrome até os 70 anos. Tendo em vista a possibilidade atual de diagnóstico molecular, metade dos portadores são identificados antes das manifestações clínicas da doença18.
Dessa maneira, a VHL é definida pela formação de vários tumores benignos e eventualmente malignos hipervascularizados, tanto em SNC como em órgãos viscerais, além de múltiplos cistos em pâncreas e rins7,18.
Assim, os critérios diagnósticos propostos para a síndrome reúnem achados clínicos e moleculares: história familiar de VHL que apresenta lesão característica da doença como hemangioblastoma do SNC ou retina ou lesão visceral (carcinoma de células renais, feocromocitoma, cistos ou tumor endócrino pancreático ou cistoadenoma do epidídimo); aqueles sem antecedentes familiares de VHL precisam possuir dois ou mais hemangioblastomas do SNC ou retina ou um hemangioblastoma associado à lesão visceral para preencher os critérios de diagnóstico4,19.
Hemangioblastomas são neoplasias vasculares benignas e os pacientes que possuem VHL têm maior chance de desenvolvê-las, principalmente no cerebelo (16-69%) e retina (49%-62%). Contudo, esse tumor também pode aparecer no tronco cerebral (5-22%), na medula espinhal (13-53%), cauda equina (11%) ou supratentorial (1-7%)20 (Tabela 1). Aparecem geralmente entre a segunda e terceira décadas de vida e muitas vezes são as primeiras manifestações da síndrome3,17. Esses tumores podem ser assintomáticos, mas também manifestar sintomas pelo efeito de massa e compressão de estruturas adjacentes, como cefaleias, náuseas e vômitos ou até déficits sensoriais e/ou motores e ataxia. Logo, estão relacionados à morbidade e à mortalidade na síndrome de VHL3,17,18. Seu diagnóstico no SNC é pela ressonância magnética com contraste e, em tumores com componentes sólidos, tem-se isossinal em T1 e hipersinal em T2, e o de componente cístico tem hipossinal em T1 e hipersinal em T25. O seu tratamento, de modo geral, envolve ressecção cirúrgica17.
O paciente reportado tinha cistos no baço. O acometimento de vísceras é uma característica marcante dos pacientes que apresentam a síndrome de VHL. Embora nesse caso não haja acometimento renal, cistos benignos e carcinomas de células claras se destacam na síndrome de VHL.
A possibilidade de desenvolver carcinoma de células renais até os 60 anos é de até 60%, sendo a principal causa de morte desses pacientes. Eles inicialmente são assintomáticos, evoluindo para quadros de hematúria, dor no flanco ou massa abdominal palpável (Tabela 1). O diagnóstico é feito mediante tomografia computadorizada contrastada. Pacientes com carcinoma de células renais podem desenvolver metástases, com predileção por osso, pulmão e fígado3. O tratamento desses pacientes envolve ressecção cirúrgica para aqueles tumores com mais de 3 cm.
Os feocromocitomas surgem de células cromafins na medula adrenal. Ocorrem em 16-20% dos pacientes e costumam aparecer na segunda década de vida17,20 (Tabela 1). Esses tumores produzem catecolaminas causando um quadro de hipertensão, taquicardia, palpitação, dores de cabeça, sudorese, palidez e náuseas, mas também podem ser assintomáticos17,18. Os estudos laboratoriais mostram excesso de catecolaminas que, em conjunto com uma imagem sugestiva, confirmam o diagnóstico20. O quadro pode causar complicações potencialmente fatais como arritmias e síndrome coronariana aguda. Dessa forma, o tratamento de escolha é cirúrgico.
Além disso, é valido relatar o aparecimento de lesões pancreáticas nos quadros de VHL que raramente sofrem malignização, além de cistoadenomas ependimários e cistoadenomas de ligamento largo17.
|
Tabela 1. Frequências das manifestações clínicas da síndrome de von Hippel-Lindau |
||||||||||||
|
||||||||||||
|
Fonte: Gatti et al.5 Legenda: SNC = sistema nervoso central. |
O paciente em questão é do subtipo 2A da classificação de síndrome de VHL, dado corroborado pela alteração genética encontrada. Não é o subtipo mais comum, sendo o mais comum o subtipo I. Na investigação do caso, o achado de hemangioblastoma de SNC levantou a suspeita inicial para a síndrome, embora o paciente não tenha os outros achados clínicos esperados – hemangioblastoma de retina e feocromocitoma (Quadro 1).
Liu et al.9 reportaram a primeira família chinesa com a mutação VHL p.Tyr98His (Y98H) amplamente considerada em pacientes com mutação do VHL do tipo 2A. Foram avaliadas quatro gerações, contabilizando um total de 15 familiares portadores da mutação em questão, dos quais apenas quatro foram diagnosticados com feocromocitomas característicos do tipo 2. O estudo também verificou que ainda há um número pequeno de pacientes portadores da mutação que apresentam carcinoma de células renais, frequentemente presente no subtipo 2A, indicando que a incompatibilidade genótipo-fenótipo não é impossível, o que é demonstrado também pelo caso descrito neste estudo9 (Quadro 1).
|
Quadro 1. Classificação da síndrome de von Hippel-Lindau de acordo com a natureza do tumor encontrado |
||||||||||||||||||
|
||||||||||||||||||
|
Fonte: Nielsen et al.6 Legendas: SNC = sistema nervoso central; VHL = von Hippel-Lindau. |
Assim, trata-se de um caso raro de VHL do tipo 2A após a identificação da variante VHL c.292T>C. Diante disso, manifestações características da síndrome, testes clínicos cuidadosos e análise de mutações podem auxiliar no diagnóstico e, de fato, confirmar a síndrome. O guideline do “VHL Alliance”21 sugere acompanhamento clínico por meio de consultas com anamnese e exame físico anualmente nos primeiros cinco anos do diagnóstico, com aferição da pressão arterial e pulso, exame de retina e metanefrinas urinárias. Exames de imagem como ressonância de cérebro, coluna e abdome total com contraste devem ser feitos a cada dois anos dos 30 aos 65 anos.
Em relação à sobrevida dos pacientes com VHL, historicamente é menor do que a da população em geral, com uma expectativa de vida de 49 anos; porém trata-se de um estudo realizado há mais de 25 anos. Contudo, um estudo retrospectivo recente com homens e mulheres dinamarqueses nascidos em 2000 com síndrome de VHL demonstrou uma expectativa de vida de 67 e 60 anos respectivamente19.
Por fim, é valido ressaltar a importância da multidisciplinaridade no cuidado do paciente com VHL, pois, em razão da alta frequência de múltiplos tumores em vários órgãos e sistemas, uma equipe multidisciplinar com um programa de reabilitação individualizado se faz necessária para melhores condições de vida22. O papel das várias especialidades médicas, como neurocirurgião, oftalmologista, urologista, geneticista, nefrologista, clínico, é crucial. Além disso, seu efeito é potencializado, visando à melhora do paciente, quando associado à fisioterapia, terapia ocupacional ou psicologia22-24. Portanto, a cronicidade e o curso da doença têm grandes implicações nas questões da vida diária, permeando aspectos sociais e funcionais. Nesse sentido, lidar com os agravos desse quadro requer apoio psicológico para lidar com questões de saúde mental geradas pelas incertezas associadas a essa doença22. Assim, questões funcionais que surgem associadas às lesões em SNC ou a remoções cirúrgicas que podem causar déficits neurológicos e comprometimento da funcionalidade devem ser abordadas por fisioterapeutas e terapeutas ocupacionais a partir de tratamento direto dos sintomas trazendo melhora máxima possível a esses pacientes24.
Assim como no caso exposto, a exérese dos tumores, causados pela VHL com características, em sua maioria, benignas ou de carregar baixa taxa de metástase, é a escolha de tratamento quando bem indicada para esses pacientes17. Como perspectivas, há terapias promissoras sob investigação direcionadas à síndrome de VHL. Elas se concentram principalmente na inibição do crescimento do tumor primário e da angiogênese. As drogas objetivam agir na história natural da doença, são anticorpos monoclonais (p. ex.: bevacizumabe, ranibizumabe) que têm resultados inconclusivos, sendo usados como adjuvantes. Inibidores da tirosina-quinase, drogas direcionadas que bloqueiam as cascatas de transdução do sinal celular, com base na via tumoral do fator de crescimento endotelial vascular, do inglês vascular endothelial growth fator (VEGF) na VHL (p. ex.: semaxanib, sunitinibe, pazopanibe, erlotinibe, dovitinib, sorafenibe), possuem resultados positivos, com melhora sintomática e até mesmo regressão de tumores. Aptâmeros de RNA, que inibem uma das isoformas de VEGF, foram testados por ensaios clínicos e em um estudo que está na Fase 1. Modificadores de resposta biológica aumentam ou modulam a resposta imune autóloga do hospedeiro à síndrome de VHL. Alguns desses modificadores, testados in vitro e em pequenos grupos de pacientes com VHL: roquinimex, talidomida, IFN-α-2a, inibidores do HIF2α, octreotide, claritromicina e imunoterapia, estão sob avaliação25.
CONCLUSÃO
O achado de hemangioblastoma em SNC ou retina, feocromocitoma ou carcinoma de células renais, associado ou não a histórico familiar positivo para a síndrome, deve levantar alta suspeita para a possibilidade do diagnóstico de síndrome de VHL. As doenças que possuem incidência rara representam um desafio no dia a dia do médico.
Todos os autores contribuíram substancialmente na concepção e/ou no planejamento do estudo; na obtenção, análise e/ou interpretação dos dados; na redação e/ou revisão crítica; e aprovaram a versão final a ser publicada.
DECLARAÇÃO DE CONFLITO DE INTERESSES
Nada a declarar.
FONTES DE FINANCIAMENTO
Não há.
REFERÊNCIAS
1. Hamosh A, Scott AF, Amberger J, et al. Online Mendelian Inheritance in Man (OMIM). Human Mutation. 2000;15(1):57-61. doi: https://doi.org/10.1002/(SICI)1098-1004(200001)15:1<57::AID-HUMU12>3.0.CO;2-G
2. Findeis-Hosey JJ, McMahon KQ, Findeis SK. Von Hippel-Lindau disease. J Pediatr Genet. 2016;5(2):116-23. doi: https://doi.org/10.1055/s-0036-1579757
3. Fujita PA, Rhead B, Zweig AS, et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011;39(Suppl 1):D876-82. doi: https://doi.org/10.1093/nar/gkq963
4. Maher ER, Sandford RN. von Hippel-Lindau disease: an Update. Curr Genet Med Rep. 2019;7:227-35. doi: https://doi.org/10.1007/s40142-019-00180-9
5. Gatti R, Pereira MAA, Giannella Neto D. Síndrome de von Hippel-Lindau. Arq Bras Endocrinol Metab. 1999;43(5):377-88. doi: https://doi.org/10.1590/S0004-27301999000500011
6. Nielsen SM, Rhodes L, Blanco I, et al. Von Hippel-Lindau disease: genetics and role of genetic counseling in a multiple neoplasia syndrome. J Clin Oncol. 2016;34(18):2172-81. doi: https://doi.org/10.1200/jco.2015.65.6140
7. Friedrich CA. Von Hippel-Lindau syndrome. A pleomorphic condition. Cancer. 1999;86(11 Suppl):2478-82. doi: https://doi.org/10.1002/(SICI)1097-0142(19991201)86:11+<2478::AID-CNCR4>3.0.CO;2-5
8. Kleihues P, Louis DN, Scheithauer BW, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61(3):215-25. doi: https://doi.org/10.1093/jnen/61.3.215
9. Liu P, Zhu F, Li M, et al. Von Hippel-Lindau “Black Forest” mutation inherited in a large Chinese family. Gland Surg. 2019;8(4):343-53. doi: https://doi.org/10.21037/gs.2019.08.03
10. Nykamp K, Anderson M, Powers M, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. 2017;19(10):1105-17. doi: https://doi.org/10.1038/gim.2017.37
11. Louise M Binderup M, Smerdel M, Borgwadt L, et al. von Hippel-Lindau disease: Updated guideline for diagnosis and surveillance. Eur J Med Genet. 2022;65(8):104538. doi: https://doi.org/10.1016/j.ejmg.2022.104538
12. Conselho Nacional de Saúde (BR). Resolução nº 466, de 12 de dezembro de 2012. Aprova as diretrizes e normas regulamentadoras de pesquisas envolvendo seres humanos. Diário Oficial da União, Brasília, DF. 2013 jun 13; Seção 1:59.
13. Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260(5112):1317-20. doi: https://doi.org/10.1126/science.8493574
14. Crossey PA, Richards FM, Foster K, et al. Identification of intragenic mutations in the Von Hippel-Lindau disease tumour suppressor gene and correlation with disease phenotype. Hum Mol Genet. 1994;3(8):1303-8. doi: https://doi.org/10.1093/hmg/3.8.1303
15. Lenglet M, Robriquet F, Schwarz K, et al. Identification of a new VHL exon and complex splicing alterations in familial erythrocytosis or von Hippel-Lindau disease. Blood. 2018;132(5):469-83. doi: https://doi.org/10.1182/blood-2018-03-838235
16. van Leeuwaarde RS, Ahmad S, Links TP, et al. Von Hippel-Lindau syndrome. In: Adam MP, Everman DB, Mirzaa GM, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993 [cited 2022 Sep 16]. Available from: https://pubmed.ncbi.nlm.nih.gov/20301636/
17. Varshney N, Kebede AA, Owusu-Dapaah H, et al. A review of Von Hippel-Lindau syndrome. J Kidney Cancer VHL. 2017;4(3):20-9. doi: https://doi.org/10.15586/jkcVHL.2017.88
18. Mikhail MI, Singh AK. Von Hippel Lindau syndrome. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 [cited 2022 abr 12]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK459242/
19. Binderup MLM, Jensen AM, Budtz-Jørgensen E, et al. Survival and causes of death in patients with von Hippel-Lindau disease. J Med Genet. 2017;54(1):11-18. doi: https://doi.org/10.1136/jmedgenet-2016-104058
20. Chittiboina P, Lonser RR. Von Hippel-Lindau disease. Handb Clin Neurol. 2015;132:139-56. doi: https://doi.org/10.1016/B978-0-444-62702-5.00010-X
21. VHL Alliance. Lo que se necesita saber sobre la enfermedad de von Hippel-Lindau: Un manual de referencia para individuos con von-Hippel-Lindau (VHL), sus familias y sus equipos médicos. 6 ed. Rev. Boston (MA): VHL Alliance; 2020 [acesso 2022 abr 12]. Disponível em: https://www.vhl.org/storage/2023/01/El-Manual-de-la-VHLA_2021-Spanish-VHL-Handbook.pdf
22. Schmid S, Gillessen S, Binet I, et al. Management of von hippel-lindau disease: an interdisciplinary review. Oncol Res Treat. 2014;37(12):761-71. doi: https://doi.org/10.1159/000369362. Erratum in: Oncol Res Treat. 2015;38(1-2):50. doi: https://doi.org/10.1159/000375284
23. Wolters WPG, Dreijerink KMA, Giles RH, et al. Multidisciplinary integrated care pathway for von Hippel-Lindau disease. Cancer. 2022;128(15):2871-9. doi: https://doi.org/10.1002/cncr.34265
24. Tsingeli P, Papadatou MC, Psillaki D, et al. Rehabilitation management in two siblings with Von Hippel-Lindau syndrome: a case series. J Musculoskelet Neuronal Interact. 2021;21(2):326-31. Cited in: PubMed; PMID 34059579.
25. Gläsker S, Vergauwen E, Koch CA, et al. Von Hippel-Lindau disease: current challenges and future prospects. Onco Targets Ther. 2020;13:5669-90. doi: https://doi.org/10.2147/OTT.S190753
Aprovado em 1/11/2022
Editora-científica: Anke Bergmann. Orcid iD: https://orcid.org/0000-0002-1972-8777
![]()
Este é um artigo publicado em acesso aberto (Open Access) sob a licença Creative Commons Attribution, que permite uso, distribuição e reprodução em qualquer meio, sem restrições, desde que o trabalho original seja corretamente citado.
©2019 Revista Brasileira de Cancerologia | Instituto Nacional de Câncer | Ministério da Saúde