RELATO DE CASO
Cordoma Sacral: Relato de uma Rara Neoplasia Maligna
Sacral Chordoma: Report of a Rare Malignant Neoplasm
Cordoma Sacro: Informe de una Rara Neoplasia Maligna
doi: https://doi.org/10.32635/2176-9745.RBC.2023v69n1.3519
Rafael Gonçalves Zimmer1; Isadora Lyrio Stábille2; Francine Ribeiro Potros3; Adriana Batista Alves Martins4
1-4Universidade Federal do Triângulo Mineiro (UFTM), Hospital de Clínicas. Uberaba (MG), Brasil.
E-mail: rgzimmer91@gmail.com. Orcid iD: https://orcid.org/0000-0002-4042-2975
E-mail: isadora.lyrio@hotmail.com. Orcid iD: https://orcid.org/0000-0003-4976-7897
E-mail: franpotros@hotmail.com. Orcid iD: https://orcid.org/0000-0001-5686-9985
E-mail: dri_amartins@hotmail.com. Orcid iD: https://orcid.org/0000-0002-0936-5474
Endereço para correspondência: Rafael Gonçalves Zimmer. Rua João Alvares Correia, 111 – apto. 91 – Vila Mariana/Chácara Klabin. São Paulo (SP), Brasil. CEP 04115-030. E-mail: rgzimmer91@gmail.com
Introdução: O cordoma é um tipo de sarcoma cuja malignidade óssea primária se origina da notocorda e se localiza no eixo espinhal entre o clivus e o sacro. A primeira descrição dessa patologia foi em 1857. Na epidemiologia da doença, são mais afetados pacientes entre 40 e 60 anos, sendo o principal sítio de acometimento a região sacrococcígea. O quadro clínico é variável conforme o local acometido com sintomas geralmente inespecíficos, gerando atrasos no diagnóstico feito por biópsia. Entre as opções de tratamento, o principal método é a ressecção cirúrgica com margens, que pode ser associada à radioterapia ou à radiocirurgia quando necessário; os sítios de metástases mais comuns são pulmões, ossos, fígado e linfonodos locais. Relato do caso: Paciente, sexo feminino, 62 anos, iniciou com quadro de dor em região coccígea com piora ao sentar-se e surgimento de lesão nodular com crescimento progressivo recebendo diagnóstico de cordoma, após biópsia da lesão, depois de três anos. Em razão da lesão extensa, optou-se inicialmente por tratamento com quimio e radioterapia para citorredução. Pela pouca responsividade, foi submetida ao tratamento de sacralectomia com sucesso, porém apresentou como complicação deiscência de ferida operatória e necessidade de reabordagem, desde então sem recorrência no seguimento clínico. Conclusão: Assim, evidencia-se a necessidade de novas pesquisas sobre o cordoma, um tumor raro e de baixa responsividade aos tratamentos não cirúrgicos, visando a melhorar a terapêutica quimioterápica dessa neoplasia potencialmente deformante.
Palavras-chave: neoplasias ósseas; cordoma; região sacrococcígea; relatos de casos.
ABSTRACT
Introduction: Chordoma is a type of sarcoma, a primary bone malignancy that originates from the notochord and is located on the spinal axis between the clivus and the sacrum. The first description of this pathology occurred in 1857. Patients between 40 and 60 years old are the most affected according to the disease’s epidemiology, the main site involved is the sacral/coccygeal region. The clinical condition is variable depending on the site affected, with generally nonspecific symptoms, delaying the diagnosis made by biopsy. Among the treatment options, surgical resection with margins is currently the main method, and may be associated with radiotherapy or radiosurgery when necessary; the most common metastatic sites are lungs, bones, liver and local lymph nodes. Case report: A 62-year-old female patient had pain in the coccygeal region, worsening while sitting and the appearance of a nodular lesion with progressive growth, diagnosed as a chordoma three years later, after biopsy of the lesion. Due to the extensive lesion, initially she was submitted to chemotherapy and radiotherapy for cytoreduction, but because of the poor response, she was successfully submitted to sacralectomy, however, dehiscence of the surgical wound was detected, and the patient underwent a new approach; since then, no recurrence in the clinical follow-up. Conclusion: Apparently, it is clear the necessity for further investigations on chordoma, a rare tumor with poor response to non-surgical treatments, in order to improve the chemotherapy for this potentially deforming neoplasm.
Key words: bone neoplasms; chordoma; sacrococcygeal region; case reports.
RESUMEN
Introducción: El cordoma es un tipo de sarcoma, una malignidad ósea primaria que se origina en la notocorda y se localiza en el eje espinal entre el clivus y el sacro. La primera descripción de esta patología fue en 1857. En la epidemiología de la enfermedad, los pacientes entre 40 y 60 años son los más afectados, siendo el principal sitio de afectación la región sacrocoxígea. El cuadro clínico es variable según el sitio afectado, con síntomas generalmente inespecíficos, lo que provoca retrasos en el diagnóstico realizado mediante biopsia. Entre las opciones de tratamiento, la resección quirúrgica con márgenes es actualmente el principal método, pudiendo asociarse a radioterapia o radiocirugía cuando sea necesario; los sitios más comunes de metástasis son los pulmones, los huesos, el hígado y los ganglios linfáticos locales. Informe del caso: Paciente, sexo femenino, de 62 años inició con dolor en la región coccígea, empeorando al sentarse y aparición de una lesión nodular con crecimiento progresivo, recibiendo diagnóstico de cordoma, luego de biopsia de la lesión, después de tres años. Debido a la extensión de la lesión optó inicialmente por tratamiento con quimio y radioterapia para citorreducción, por la poca reactividad fue sometida con éxito al tratamiento de sacralectomía, pero presentó como complicación dehiscencia de la herida quirúrgica y necesidad de reabordaje. Desde entonces sin recurrencia en el seguimiento clínico. Conclusión: Por lo tanto, es evidente la necesidad de seguir investigando sobre el cordoma, un tumor poco frecuente con escasa respuesta a los tratamientos no quirúrgicos, con el fin de mejorar la terapia de quimioterapia para esa neoplasia potencialmente deformante.
Palabras clave: neoplasias óseas; cordoma; región sacrococcígea; informes de casos.
INTRODUÇÃO
Cordoma é um tipo de sarcoma cuja malignidade óssea primária se origina da notocorda e se localiza no eixo espinhal entre o clivus e o sacro1-6. A primeira descrição dessa patologia foi em 1857, quando foram observadas células vacuoladas ao microscópio e se instituiu o termo physaliphorous (derivado da palavra grega para bolhas). Em 1890, passou a se chamar cordoma, associado à hipótese da origem da notocorda2. Na epidemiologia dessa doença, são mais afetados pacientes entre 40 e 60 anos. O principal sítio de acometimento (50%) é a região sacrococcígea com o restante acometendo crânio e corpos vertebrais1,6,7.
O quadro clínico varia conforme o local acometido. Os sintomas iniciais são geralmente inespecíficos, o que contribui para atraso no diagnóstico2. Este é feito com a aspiração por agulha fina ou biópsia por agulha grossa. Visto que há risco de disseminação tumoral, deve-se proceder com cautela e excisar o tumor completamente2. A braquiúria é um fator de transcrição considerado marcador diagnóstico sensível e específico para esse tumor6. Essa afecção oncológica faz diagnóstico diferencial com múltiplas entidades benignas e malignas, por isso sua importância.
Entre as opções de tratamento, a principal é a ressecção cirúrgica com margens, que pode ser associada à radioterapia ou à radiocirurgia se necessário2. Os sítios de acometimento metastático mais comuns são os pulmões, ossos, fígado e linfonodos locais6, não identificados no caso relatado. Sabe-se, ainda, que existem quatro subtipos histológicos de acordo com a classificação da Organização Mundial da Saúde (OMS) de cordomas: o clássico/convencional, condroide, indiferenciado e o pouco diferenciado6,8. Entre eles, o cordoma clássico é o mais comum6.
Este artigo se propõe a reafirmar a necessidade de mais pesquisas sobre a patologia, bem como novas terapias ou alternativas adicionais de tratamento buscando melhorar a resposta terapêutica e reduzir medidas invasivas e suas complicações.
O presente relato foi aprovado pelo Comitê de Ética em Pesquisa (CEP) do Hospital de Clínicas da Universidade Federal do Triângulo Mineiro (UFTM), sob número de parecer 5.399.200 (CAEE: 55707222.3.0000.8667), em conformidade com as diretrizes da Resolução do Conselho Nacional de Saúde (CNS) n.º 466, de 12 de dezembro de 20129.
RELATO DO CASO
Paciente, sexo feminino, 62 anos, divorciada e educadora física aposentada. Apresentou quadro progressivo de dor em região coccígea com piora ao se sentar-se iniciada em meados de 2018. Previamente sem comorbidades, exceto acompanhamento por osteoartrose e clipagem de aneurisma cerebral; ex-tabagista (15 anos-maço) sem história de etilismo.
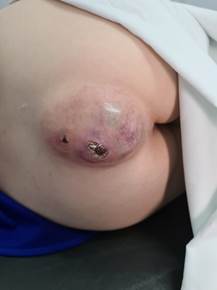
Em 2021, buscou atendimento ortopédico ao sentir piora acentuada da dor sacrococcígea sem irradiação e crescimento da lesão no último ano associado à perda ponderal de quatro quilos em seis meses, apresentando tumoração de aproximadamente 10 cm no sacro com lesão ulcerada (Figura 1). Foi feita a solicitação de exames para estadiamento (tomografias e cintilografia), contraindicando a ressonância magnética em função do clipe metálico no sistema nervoso central (SNC), chegando-se à hipótese de cordoma.
|
|
|
Figura 1. Lesão tumoral ulcerada com 10 cm aproximadamente. Uberaba (2020) |
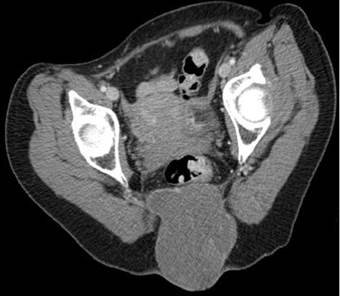
A paciente passou por avaliação da coloproctologia a respeito do caso, porém, visto que a massa não apresentava acometimento gastrointestinal em tomografia de abdome total (volumosa massa com realce heterogêneo e contornos lobulados, de limites definidos, centrada no cóccix, promovendo sua destruição, medindo cerca de 11,2 x 10,5 x 7,8 (Figura 2), estendendo-se para a região pressacral, sem plano de clivagem com o reto baixo e para a região posterior, sem plano de clivagem com a pele, ficando restrita à parte óssea), foi indicado apenas rastreio oncológico para neoplasia colorretal em momento oportuno.
|
|
|||
|
Figura 2. Tomografia computadorizada de sacro revelando a lesão tumoral 11,2 x 10,5 x 7,8 cm |
Posteriormente, realizou-se biópsia da lesão (neoplasia maligna com células com citoplasma "em bolhas"/células fisalíferas arranjadas em lóbulos e em meio à abundante matriz mixoide), confirmando o diagnóstico de cordoma. A cintilografia demonstrou hipercaptação de L3/L4/L5; tomografias de tórax e abdome sem evidências de linfonodomegalias ou metástases para esses locais. Por se tratar de lesão extensa e inicialmente inoperável, a paciente foi encaminhada para quimio e radioterapia.
Iniciou o esquema quimioterápico com imatinib 800 mg/dia (Glivec), além de radioterapia externa conformacional na dose de 50Gy/25 frações sobre a região sacral, em março de 2021. Após dois meses de tratamento, fez-se uma nova tomografia da região pélvica, agora com discreta redução das dimensões previamente apresentadas. Durante o tratamento, apresentou os seguintes efeitos colaterais: edema palpebral, diarreia e incontinência fecal.
Por causa da pequena resposta ao tratamento, apesar dos riscos, optou-se por abordagem cirúrgica conjunta no campo operatório – ortopedia e cirurgia do aparelho digestivo (CAD) – para sacralectomia que seria realizada pela ortopedia, porém com auxílio da equipe da CAD em função do tamanho da ressecção e do risco de lesão do retossigmoide. A cirurgia foi realizada em 28/6/21, após suspensão do imatinib. Depois do procedimento, houve evolução com deiscência de ferida operatória com necessidade de desbridamento, ressutura e posterior realização de retalho miocutâneo (Figura 3).
|
|
|
Figura 3. Resultado pós-ressecção e retalho miocutâneo. Uberaba (2021) |

Apresentando boa evolução na cicatrização da ferida cirúrgica, foi reavaliada quanto à possibilidade de recidiva local e, até o momento, encontra-se sem sinais de novos crescimentos. Um novo rastreio radiológico foi feito e o seguimento oncológico mantido desde então.
DISCUSSÃO
Os cordomas são tumores com caráter insidioso e crescimento lento que surgem no corpo vertebral, podendo acometer a medula espinhal ou nervos e tecidos moles paraespinhais3,6,7. A paciente iniciou com sintomas há aproximadamente quatros anos do momento do diagnóstico. Houve piora ao longo dos anos, ratificando as características de sua evolução relatadas neste trabalho.
Relatos da literatura mostram que as metástases são poucos frequentes e correspondem a 30% dos cordomas sacrococcígeos5-7. Neste caso, não houve lesões secundárias visualizadas no rastreio.
Em relação aos exames de imagem, na tomografia computadorizada pode se observar destruição óssea lítica e/ou uma massa de tecido mole. Por vezes, em 30% a 70% dos casos evidencia-se calcificação7. Na ressonância magnética, as imagens ponderadas em T1 mostram os cordomas isotensos ou levemente hipointensos em comparação com o músculo e, nas imagens em T2, hiperintensos ao músculo. Na cintilografia óssea, cordomas podem apresentar absorção reduzida ou distribuição normal do isótopo7. No presente caso, houve hipercaptação de L3/L4/L5 à cintilografia; ressecções radicais em bloco ainda são o tratamento de escolha, associado a fótons de alta energia ou radiação de feixe de prótons para doença recorrente ou residual7. No entanto, pelo tamanho da lesão tumoral da paciente e os riscos de complicações inerentes ao procedimento, inicialmente optou-se por ciclos de quimioterapia e radioterapia para redução do volume tumoral com o objetivo de facilitar a ressecção. Terapias sistêmicas são indicadas no cordoma no caso de evidência de doença progressiva avançada incontrolável com cirurgia ou radioterapia associada a sintomas clínicos8.
Nos tumores sacrais, há uma complexa anatomia da região tornando as ressecções tecnicamente difíceis. Abordagens unidirecionais são frequentemente combinadas para obter uma exposição adequada. Não obstante, pode haver complicações como perda das funções intestinais, vesicais e sexuais como consequência da cirurgia4,7. A paciente do relato manifestou sintomas de incontinência urinária, fecal e liberação de flatos com necessidade de fisioterapia para reabilitação.
A quimioterapia citotóxica que apresenta resposta satisfatória ao tratamento de sarcomas avançados de tecidos moles e ósseos é pouco relevante no cordoma5,8. Atualmente, tem-se utilizado o inibidor de tirosina-quinase imatinib, em função da crescente identificação de alvos moleculares potencialmente farmacológicos, como o receptor beta do fator de crescimento derivado de plaquetas (PDGF), o fosfoinositídeo 3-quinase (PI3K/mTOR), o receptor do fator de crescimento epidérmico (EGFR), o fator de crescimento endotelial vascular (VEFG) e a tirosina-proteína quinase Met (PTKs-MET). Entretanto, a resposta a todos esses agentes é muito baixa no cordoma8. Neste caso, foi utilizado o imatinib (Glivec), com o dobro da dose habitual durante dois meses, sem alcançar resposta satisfatória, sendo interrompido o uso para programação cirúrgica.
Esses tumores são considerados resistentes à radiação, entretanto alguns estudos relatam que a excisão subtotal junto à radioterapia foi superior à excisão subtotal sozinha no prolongamento da sobrevida livre de doença. A dose mínima eficiente varia entre 60 e 65 Gy7. É considerada uma resposta clínica satisfatória a redução de pelo menos 30% no diâmetro máximo do tumor3. No presente caso, após dois meses do tratamento clínico, houve uma redução discreta relatada, o que demonstra ineficiência, apesar de a dose aplicada na terapêutica ter sido inferior às experiências previamente relatadas na literatura. Optou-se pela dose abaixo do convencional pelo receio de gerar dano locorregional intestinal pela extensão/proximidade ao intestino.
A ressecção cirúrgica em blocos é considerada padrão-ouro no tratamento dessa patologia6. No presente caso, não havia comprometimento do tubo digestório, sendo a presença de margens livres na ressecção cirúrgica um importante preditor prognóstico e taxa de recorrência local e a distância3,4,6,10. Complicações pós-operatórias não são raras, variando de 40% a 60% dos casos, e incluem infecção do sítio cirúrgico, deiscência da ferida operatória e presença de hérnias sacrais4,10. No pós-operatório, a paciente em questão apresentou deiscência de ferida operatória com necessidade de desbridamento, ressutura e posterior realização de retalho miocutâneo. Isso demonstrou a grande morbidade relacionada ao quadro e às múltiplas complicações que podem surgir após o procedimento cirúrgico. Dessa forma, pacientes com essa neoplasia e com necessidade de abordagem cirúrgicas frequentemente necessitam de longas internações hospitalares e múltiplas reintervenções para resolvê-las10.
CONCLUSÃO
Diante do caso, o cordoma mostra-se um tumor raro sem muitas informações adicionais na literatura. As opções de tratamento disponíveis e realizadas na paciente e sua baixa responsividade aos tratamentos não cirúrgicos torna evidente a importância de novas pesquisas com o objetivo de melhorar a proposta terapêutica quimioterápica dessa neoplasia potencialmente deformante, a fim de evitar que novos pacientes passem por complicações muito comuns oriundas da abordagem cirúrgica. Dessa forma, o presente relato serve para reafirmar a necessidade de mais pesquisas, possibilitando a futuros pacientes menos morbidade e reduzindo internações e custos.
AGRADECIMENTOS
Ao Prof. Dr. Adriano Jander Ferreira, médico-ortopedista no Hospital de Clínicas da UFTM, por ter cedido as imagens do procedimento cirúrgico que enriqueceram este relato.
Rafael Gonçalves Zimmer e Isadora Lyrio Stábille contribuíram substancialmente na concepção e/ou no planejamento do estudo; na análise e/ou interpretação dos dados; na redação e/ou revisão crítica. Francine Ribeiro Potros e Adriana Batista Alves Martins contribuíram na redação e/ou revisão crítica. Todos os autores aprovaram a versão final a ser publicada.
DECLARAÇÃO DE CONFLITO DE INTERESSES
Nada a declarar.
FONTES DE FINANCIAMENTO
Não há.
REFERÊNCIAS
1. Tenny S, Varacallo M. Chordoma. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2022.
2. Williams BJ, Raper DM, Godbout E, et al. Diagnosis and treatment of chordoma. J Natl Compr Canc Netw. 2013;11(6):726-31. doi: https://doi.org/10.6004/jnccn.2013.0089
3. Atalar H, Selek H, Yildiz Y, et al. Management of sacrococcygeal chordomas. Int Orthop. 2006;30(6):514-8. doi: https://doi.org/10.1007/s00264-006-0095-x
4. Aguiar Junior S, Andrade WP, Baiocchi G, et al. Natural history and surgical treatment of chordoma: a retrospective cohort study. São Paulo Med J. 2014;132(5):297-302. doi: https://doi.org/10.1590/1516-3180.2014.1325628
5. Hindi N, Casali PG, Morosi C, et al. Imatinib in advanced chordoma: a retrospective case series analysis. Eur J Cancer. 2015;51(17):2609-14. doi: https://doi.org/10.1016/j.ejca.2015.07.038
6. Scheipl S, Igrec J, Leithner A, et al. [Chordoma: is there a molecular basis for diagnosis and treatment?] Pathologe. 2020;41(2):153-62. German. doi: https://doi.org/10.1007/s00292-020-00761-4
7. Sciubba DM, Chi JH, Rhines LD, et al. Chordoma of the spinal column. Neurosurg Clin N Am. 2008;19(1):5-15. doi: https://doi.org/10.1016/j.nec.2007.09.006
8. Frezza AN, Botta L, Trama A, et al. Chordoma: uptade on disease, epidemiology, biology and medical therapies. Curr Opin Oncol. 2019;31(2):114-20. doi: https://doi.org/10.1097/CCO.0000000000000502
9. Conselho Nacional de Saúde (BR). Resolução nº 466, de 12 de dezembro de 2012. Aprova as diretrizes e normas regulamentadoras de pesquisas envolvendo seres humanos. Diário Oficial da União, Brasília, DF. 2013 jun 13; Seção 1:59.
10. García-Ortega DY, Clara-Altamirano MA, Gómez-Pedraza A, et al. Tumores primarios de sacro: análisis de resultados y complicaciones. Acta ortop. Mex. 2018;32(6):354-7. doi: https://doi.org/10.35366/85433
Recebido em 7/12/2022
Aprovado em 1/2/2023
Editora-científica: Anke Bergmann. Orcid iD: https://orcid.org/0000-0002-1972-8777
![]()
Este é um artigo publicado em acesso aberto (Open Access) sob a licença Creative Commons Attribution, que permite uso, distribuição e reprodução em qualquer meio, sem restrições, desde que o trabalho original seja corretamente citado.
©2019 Revista Brasileira de Cancerologia | Instituto Nacional de Câncer | Ministério da Saúde