Gastrointestinal Stromal Tumor in a Patient with Neurofibromatosis Type I: Diagnosis after Intestinal Occlusion Secondary to Adhesions after an Episode of Pancreatitis
Tumor Estromal Gastrointestinal em Paciente com Neurofibromatose Tipo I: Diagnóstico após Oclusão Intestinal Secundária a Aderências após Episódio de Pancreatite
Tumor del Estroma Gastrointestinal en Paciente con Neurofibromatosis Tipo I: Diagnóstico tras Oclusión Intestinal Secundaria a Adherencias tras Episodio de Pancreatitis
doi: https://doi.org/10.32635/2176-9745.RBC.2023v69n2.3720
Pamela Viana e Silva1; Thiago Menezes Costa2; Renanna Najara Veras Rodrigues3; Carlos Anselmo Lima4
1-4Universidade Federal de Sergipe, Hospital Universitário, Empresa Brasileira de Serviços Hospitalares (UFS/HU/EBSERH). Aracaju (SE), Brazil. E-mails: pamelavianaesilva@gmail.com; tmc.onco@yahoo.com.br; renanna@hotmail.com; ca.lima01@gmail.com. Orcid iD: https://orcid.org/0000-0002-6645-7367; Orcid iD: https://orcid.org/0000-0003-3536-6021; Orcid ID: https://orcid.org/0000-0002-7131-6403; Orcid iD: https://orcid.org/0000-0003-4269-7320
Corresponding author: Pamela Viana e Silva. Rua João Gêniton da Costa, 246, Bloco 10, Apto. 204 – Jabotiana. Aracaju (SE), Brazil. CEP 49095-796. E-mail: pamelavianaesilva@gmail.com
ABSTRACT
Introduction: Gastrointestinal stromal tumors (GIST), although relatively rare, account for 80% of mesenchymal tumors of the digestive tract. They manifest in any part of the alimentary tract and are derived from Cajal cells. They may occur sporadically or be associated with familial syndromes such as neurofibromatosis type I. The clinical picture is variable, and they are often diagnosed incidentally. The diagnosis requires imaging tests associated with histopathological and immunohistochemical analysis. The best strategy for treatment is surgical resection and cases should be analyzed individually to verify additional advantages with the association of systemic therapy. This study aims to present an unusual case of GIST associated with neurofibromatosis type I in a patient with incidental diagnosis after semi-intestinal occlusion secondary to an episode of pancreatitis, in addition to performing a literature review on the subject. Case report: A 49-year-old woman with a history of severe pancreatitis presented with intestinal obstruction approximately 8 months after this episode. Abdominal computed tomography revealed a heterogeneous formation in the mesogastric region, measuring 6.6 x 5.1 x 5.3 cm. She underwent surgical resection and histopathological and immunohistochemical studies confirmed the diagnosis of GIST. Six months after diagnosis, the patient is in good general condition and is on systemic therapy. Conclusion: GIST are rare tumors, but their diagnosis should come to mind in patients with neurofibromatosis type 1 with abdominal masses.
Key words: gastrointestinal stromal tumors; neurofibromatosis 1; gastrointestinal neoplasms.
RESUMO
Introdução: Os tumores do estroma gastrointestinal (GIST), embora relativamente raros, correspondem a 80% dos tumores mesenquimais do trato digestivo. Manifestam-se em qualquer parte do trato alimentar e são derivados das células de Cajal. Podem ocorrer de forma esporádica ou associados a síndromes familiares como a neurofibromatose tipo I. O quadro clínico é variável, sendo frequentemente diagnosticados de forma incidental. O diagnóstico requer realização de exames de imagem associados à análise histopatológica e imuno-histoquímica. A melhor estratégia para o tratamento é a ressecção cirúrgica e os casos devem ser analisados individualmente para verificar vantagens adicionais com a associação da terapia sistêmica. O objetivo deste trabalho é apresentar um caso incomum de GIST associado à neurofibromatose tipo I em uma paciente com diagnóstico incidental após semioclusão intestinal secundárias a episódio de pancreatite, além de realizar revisão de literatura sobre o assunto. Relato do caso: Mulher, 49 anos de idade, com passado de pancreatite grave, apresentou quadro de oclusão intestinal cerca de oito meses após esse episódio. A tomografia computadorizada de abdome revelou formação heterogênea em região mesogástrica, medindo 6,6 x 5,1 x 5,3 cm. Foi submetida à ressecção cirúrgica, e os estudos histopatológico e imuno-histoquímico corroboraram o diagnóstico de GIST. Seis meses após o diagnóstico, a paciente encontra-se em bom estado geral e em uso de terapia sistêmica. Conclusão: Os GIST são tumores raros, porém seu diagnóstico deve ser lembrado em pacientes com neurofibromatose tipo 1 apresentando massas abdominais.
Palavras-chave: tumores do estroma gastrointestinal; neurofibromatose 1; neoplasias gastrointestinais.
RESUMEN
Introducción: Los tumores del estroma gastrointestinal (GIST), aunque relativamente raros, representan el 80% de los tumores mesenquimales del tubo digestivo. Se manifiestan en cualquier parte del tubo digestivo y se derivan de las células de Cajal. Pueden presentarse de forma esporádica o asociarse a síndromes familiares como la neurofibromatosis tipo I. El cuadro clínico es variable y con frecuencia su diagnóstico es incidental. El diagnóstico requiere pruebas de imagen asociadas al análisis histopatológico e inmunohistoquímico. La mejor estrategia de tratamiento es la resección quirúrgica y los casos deben analizarse individualmente para verificar ventajas adicionales con la asociación de terapia sistémica. El objetivo de este trabajo es presentar un caso inusual de GIST asociado a neurofibromatosis tipo I en un paciente con diagnóstico incidental tras una semioclusión intestinal secundaria a un episodio de pancreatitis, además de revisar la literatura sobre el tema. Informe del caso: Mujer de 49 años, con antecedente de pancreatitis severa, presentó oclusión intestinal aproximadamente ocho meses después de este episodio. La tomografía computarizada de abdomen reveló una formación heterogénea en la región mesogástrica, que medía 6,6 x 5,1 x 5,3 cm. Se le realizó resección quirúrgica y los estudios histopatológicos e inmunohistoquímicos corroboraron el diagnóstico de GIST. Seis meses después del diagnóstico, la paciente se encuentra en buen estado general y en tratamiento sistémico. Conclusión: Los GIST son tumores raros, pero su diagnóstico debe considerarse en pacientes con neurofibromatosis tipo 1 que presentan masas abdominales.
Palabras clave: tumores del estroma gastrointestinal; neurofibromatosis 1; neoplasias gastrointestinales.
INTRODUCTION
Gastrointestinal stromal tumors (GIST) correspond to 1% of gastrointestinal neoplasms1,2. Although rare, they represent 80% of mesenchymal tumors of the digestive tract3-7 and 5% of all sarcomas. They can occur in any part of the alimentary tract, originating from the cells of Cajal, in the gastrointestinal myenteric plexus2-4,6,8. They are more common in the stomach (50-70%) and small intestine (20-30%), less frequently affecting the colon and rectum (2-5%) or other abdominal cavity organs such as pancreas, gallbladder, mesentery, omentum and peritoneum3,4,7.
Neurofibromatosis type I (NF1) has an autosomal dominant inheritance caused by mutations of the NF1 gene, located on chromosome 17q11.28,9. It is characterized by a wide range of manifestations and may be present in different parts of the body, including subcutaneous, bone, nervous and soft tissues2,9,10. Presumptive diagnosis is made through clinical criteria, and identification of the three main signs: neurofibromas, café au lait spots, and Lisch nodules, occurring in more than 90% of patients until puberty10.
Genetic disorders involved in NF1 is forty-five-fold more likely to develop a GIST2, in addition to other benign and malignant neoplasms, including neurofibroma, optic pathway glioma, malignant peripheral nerve sheath tumors, neuroblastoma, pheochromocytoma, and breast cancer5.
The clinicopathological features of GIST associated with NF1 are different from those of sporadic GIST. Patients are younger (mean age 49 years), lesions are multiple, occur primarily in the small intestine, are smaller in size, have low mitotic activity, and rarely exhibit mutations in the CD 117 (KIT) and Platelet-Derived Growth Factor Receptor Alpha (PDGFRA) genes. These patients lack specific mutations for GIST, therefore, they show a variable and usually incomplete response to treatment with reverse transcriptase inhibitors6,11.
Recently, specific treatments for rare subgroups of GIST, such as those associated with NF1, have appeared in the literature, contributing to the improvement both of diagnosis and management of this condition12. This study aims to present an unusual case of GIST associated with NF1 in a patient with an incidental diagnosis after adhesions secondary to an episode of pancreatitis, resulting in intestinal occlusion, in addition to literature review to contribute to the current literature.
The study complied with Resolution 466/201213 of the National Health Council and was initiated after approval by the Institutional Review Board of “Universidade Federal de Sergipe”, CAAE (Submission for Ethical Review) number 63819322.9.0000.5546, and report number 5793112.
CASE REPORT
A 49-year-old woman presented to the University Hospital of “Universidade Federal de Sergipe”/EBSERH, Aracaju, with abdominal pain associated with vomiting, constipation and weight loss of 12 kg. She had an acute necrohemorrhagic pancreatitis eight months before and abdominal CT scan demonstrated a solid lesion in the left epigastrium. She was referred to the surgical department for further investigation. The patient turned asymptomatic after clinical measures initiated at the hospital of origin. Neurofibromas and café au lait spots were observed on the entire body surface (Figure 1).
|
|
|
Figure 1. Neurofibromas and café au lait spots |

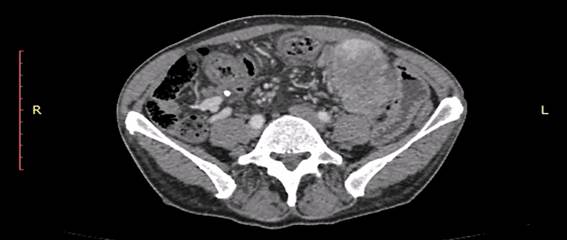
The abdomen was flat, soft, normal to percussion, and painless. A new CT scan was acquired and showed a heterogeneous formation in the left anterior region of the mesogastrium, in close relationship to the left rectus abdominis muscle and small bowel, with intermingled hypodense areas, measuring 6.6 x 5.1 x 5.3 cm (volume of 92.7 cm3) (Figure 2).
|
|
|
Figure 2. Computed tomography showing heterogeneous formation in the left anterior abdomen |
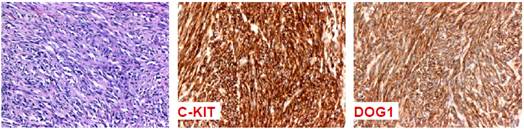
Findings during exploratory laparotomy confirmed a mass adhered to the wall of jejunum and adhesions causing partial obstruction. The patient underwent partial intestinal resection and reconstruction with end-to-end anastomosis, and resection of a skin nodule. Macroscopy described a tumor measuring 7.0 x 5.1 x 4.6 cm adhered to the surface of the segment. Histological examination showed a low-grade fusocellular neoplasm, predominantly infiltrating, apparently originating from the muscular layer. Necrosis was absent, and portrayed six mitoses per 10 high-power fields. The skin lesion was characterized as a neurofibroma. Immunohistochemical analysis showed expression of CD 117 (c-KIT), antibody discovered in GIST1 (DOG1), and absence of expression of desmin, corroborating the diagnosis of gastrointestinal stromal tumor (Figure 3).
|
|
|
Figure 3. Immunohistochemical study characterizing positivity for CD 117 (c-KIT)
|
The patient evolved uneventfully and was discharged on the fifth postoperative day. After 14 months of follow-up, she remains in good general health, using Imatinib 400 mg/day.
DISCUSSION
GIST patients develop variable and nonspecific clinical presentation, according to size and location of the tumor, but are often asymptomatic and end up being diagnosed incidentally2,3,14 as in the present case. The most frequent symptoms are anemia, weight loss, gastrointestinal bleeding, abdominal pain, symptoms related to mass effect, and may also present intestinal obstruction, perforation, and peritonitis14.
Cells of Cajal and GIST share common markers for CD 117 (also called the KIT gene) and a calcium-activated chloride channel called DOG8. KIT is a transmembrane tyrosine kinase receptor, responsible for several cellular functions, including proliferation, adhesion, apoptosis, and cell differentiation. In GIST, mutation in the KIT gene is responsible for inappropriate activation of KIT, which causes oncogenic signaling in the cell without opposition to cell proliferation7.
Other mutations can occur affecting succinate dehydrogenase (SDH), BRAF, KRAS, and NF1. Mutations in NF1 lead to formation of GIST in patients with NF18. While the two conditions may seem distinct, evidence suggests that NF1 gene may lead to increased risk of GIST. Some literature reports suggest that patients with NF1 should be monitored for GIST, especially if abdominal pain, vomiting, or gastrointestinal bleeding15,16 have been reported. Individuals having mutation in the NF1 have a probability of GIST of 7%. Conversely, NF1-associated GIST cases account for only 5% of all GIST17. The molecular mechanism associating NF1 and GIST is still not completely clear. Such cases may be due to activation of the RAS/RAF/MAP kinase signaling pathway through loss of function of the NF1 tumor suppressor gene18. As for treatment, some authors claim that early detection and management of GIST in patients with NF1 can improve their outcomes19.
GIST has been classified as to its risk of recurrence after resection, based on two prognostic factors: tumor size and mitotic count2-4,7,8. The present case high-risk classification justifies adjuvant treatment with Imatinib.
Imaging techniques can be applied to help the diagnosis. On CT, these tumors are encapsulated and have heterogeneous contrast uptake due to the presence of areas of necrosis8. It is also possible to identify areas of hemorrhage, intralesional calcification, and metastatic disease4,14. Metastases are not rare and the most affected sites include the liver and peritoneum2,3,8. Magnetic Resonance Imaging can demonstrate pelvic, mesenteric and peritoneal extension3,4,8,14.
Positron Emission Tomography improves diagnostic accuracy of metastases, and monitors clinical response to pharmacological treatment. It can detect lesions of up to 1 cm and epiploic metastases that go unnoticed on CT4. It also provides information on metabolic activity and allows the quantification of the degree of malignancy, as greater the uptake of glucose by the tumor, greater is the metabolic activity and the aggressiveness8.
On endoscopic examination, they appear as smooth submucosal tumors that protrude extrinsically into the visceral lumen and can sometimes ulcerate. Its location in the third and fourth layers (submucosa and muscularis propria) turns endoscopic biopsies inconclusive8.
Endoscopic ultrasound identifies the layers of the digestive tract with great precision, allowing accurate diagnosis of submucosal lesions. However, it does not distinguish between GIST and another neoplasm of the intestinal wall, such as leiomyoma. Thus, fine needle aspiration is recommended for the diagnosis of the lesion4. Preoperative biopsy is not mandatory in suspected GIST, but it avoids the need for lymphadenectomy, which would be crucial for neuroendocrine tumors or intestinal adenocarcinoma8. In addition, it is useful when the submucosal lesion is indeterminate or there is evidence of metastatic disease, and palliative treatment is considered4,6.
Histopathological analysis may reveal the presence of fusiform (70%), epithelioid (20%) or mixed (10%) cells3,4,14. Unlike other mesenchymal tumors, GIST expresses CD 117 antigen, demonstrated by immunohistochemical analysis14.
The best treatment strategy is surgical resection. Routine lymph node resection is not performed, since metastases are rare2-4,7,8. Lesions with suspected invasion of adjacent organs should be treated by radical monobloc resection7. Meticulous surgical technique is required to prevent tumor/capsule rupture resulting in dissemination and worst prognosis7,8.
Systemic therapy is indicated in case of metastases, non-resectable, or locally advanced tumors2,3,8,14. Imatinib, the best-studied systemic agent, is an oral inhibitor of tyrosine kinase, c-KIT receptor and PDGFRA receptor2,14. In general, the presence of KIT mutation is highly associated with response to this agent8.
In the present case, the patient was symptomatic and presented some characteristics that have been associated with GIST in patients with NF1, such as younger age, and a localized isolated lesion in the small intestine, showing high mitotic activity, upon histological evaluation demonstrated specific mutations of sporadic GIST, contrary to what has been reported in the literature. Markers KI-67, DOG-1, CD-34 and CD-117 (C-KIT) were positive. Preoperative biopsy was not performed because the lesion was considered resectable, and suspicion of GIST was high.
CONCLUSION
GIST are rare tumors, with nonspecific symptoms, but their diagnosis should always be kept in mind when evaluating patients with NF1 and abdominal masses. The ideal treatment involves surgical resection with free margins and the use of Imatinib should be individualized.
CONTRIBUTIONS
Pamela Viana e Silva and Carlos Anselmo Lima contributed to the study design, in addition to data collection and critical analysis of the results. Pamela Viana e Silva, Thiago Menezes Costa, and Renanna Najara Veras Rodrigues contributed to the wording of the manuscript. All the authors revised and approved the final version for publication.
DECLARATION OF CONFLICT OF INTEREST
There is no conflict of interests to declare.
FUNDING SOURCES
None.
REFERENCES
1. Morales HFL, Vega FAS, Diaz MEK, et al. GIST duodenal asociado a hemorragia digestiva alta. Reporte de caso. Rev Cirurgia. 2021;73(2):212-6. doi: https://doi.org/10.35687/s2452-45492021002861
2. Grezzana-Filho TJM, Mendonça TB, Golbspan L, et al. GISTs múltiplos em neurofibromatose tipo 1: diagnóstico incidental em paciente com abdome agudo. ABCD Arq Bras Cir Dig. 2009;22(1):65-8. doi: https://doi.org/10.1590/S0102-67202009000100015
3. Pinilla-Lizarraga R, Claros-Beltrán N, Mayte-Arze G. Neoplasia fusocelular - tumor de GIST: presentación de un caso. Cuad Hosp Clín. 2020;61(2):51-7.
4. Vargas-Ávila AL, Reyes-García VG, Torres-Silva C, et al. GIST en segunda porción de duodeno, abordaje quirúrgico, reporte de caso y revisión de literatura. Cir Gen. 2019;41(3):191-201.
5. Gopie P, Mei L, Faber AC, et al. Classification of gastrointestinal stromal tumor syndromes. Endocr Relat Cancer. 2018;25(2):R49-R58. doi: https://doi.org/10.1530/ERC-17-0329
6. Kayser DFL, Amaral LNF, Andrade LPC, et al. Jejunal stromal tumor and neurofibromatosis. J Coloproctology (Rio J.). 2019;39(4):385-8. doi: https://doi.org/10.1016/j.jcol.2019.05.006
7. Linhares E, Valadão M. Atualização em GIST. Rev Col Bras Cir. 2006;33(1):51-4. doi: https://doi.org/10.1590/S0100-69912006000100012
8. Townsend Jr CM, Beauchamp RD, Evers BM, et al., editors. Sabiston textbook of surgery: the biological basis of modern surgical practice. 20th ed. Philadelphia: Elsevier; 2016.
9. Shang L, Fang Z, Liu J, et al. Case report of ascending colon cancer and multiple jejunal GISTs in a patient with neurofibromatosis type 1 (NF1). BMC Cancer. 2019;19(1):1196. doi: https://doi.org/10.1186/s12885-019-6375-9
10. Moraes FS, Santos WEM, Salomão GH. Neurofibromatosis type I. Rev Bras Oftalmol. 2013;72(2):128-31. doi: https://doi.org/10.1590/S0034-72802013000200013
11. Nishida T, Tsujimoto M, Takahashi T, et al. Gastrointestinal stromal tumors in Japanese patients with neurofibromatosis type I. J Gastroenterol. 2016;51:571-8. doi: https://doi.org/10.1007/s00535-015-1132-6
12. Dantas RCM, Fernandes LFQ, Carvalho IT, et al. Coexistência de tumores estromais gastrointestinais (GISTs), feocromocitoma e paragangliomas em uma paciente com neurofibromatose tipo 1: relato de caso. Res Soc Dev. 2022;11(3):e3911326166. doi: https://doi.org/10.33448/rsd-v11i3.26166
13. Conselho Nacional de Saúde (BR). Resolução nº 466, de 12 de dezembro de 2012. Aprova as diretrizes e normas regulamentadoras de pesquisas envolvendo seres humanos. Diário Oficial da União, Brasília, DF. 2013 jun 13; Seção 1:59.
14. Muñoz-Cedeño RG, Santillán-López W, Paullán-Sani V, et al. Tumor de estroma gastrointestinal extraintestinal gigante: reporte de un caso y revisión de bibliografía. Rev Colomb Gastroenterol. 2021;36(4):532-8. doi: https://doi.org/10.22516/25007440.649
15. Hudgi AR, Azam M, Masood M, et al. The GIST of it: a rare presentation of neurofibromatosis type I. Cureus. 2021;13(6):e16034. doi: https://doi.org/10.7759/cureus.16034
16. Valencia E, Saif MW. Neurofibromatosis type 1 and GIST: Is there a correlation? Anticancer Res. 2014;34(10):5609-12.
17. Girma T, Nureta TH, Abebe DM. Unusual presentation of GIST associated with type 1 neurofibromatosis: a case report. Int J Surg Case Rep. 2023;105:107992. doi: https://doi.org/10.1016/j.ijscr.2023.107992
18. Judson I, Bulusu R, Seddon B, et al. UK clinical practice guidelines for the management of gastrointestinal stromal tumours (GIST). Clin Sarcoma Res. 2017;7:6. doi: https://doi.org/10.1186/s13569-017-0072-8
19. Abu-Abaa M, Abdulsahib A, Kananeh S, et al. A rare association between gastrointestinal stromal tumor and neurofibromatosis type 1: a case report. Cureus. 2023;15(1):e34148. doi: https://doi.org/10.7759/cureus.34148
Recebido em 1/2/2023
Aprovado em 11/5/2023
Scientific-Editor: Anke Bergmann. Orcid iD: https://orcid.org/0000-0002-1972-8777
![]()
Este é um artigo publicado em acesso aberto (Open Access) sob a licença Creative Commons Attribution, que permite uso, distribuição e reprodução em qualquer meio, sem restrições, desde que o trabalho original seja corretamente citado.
©2019 Revista Brasileira de Cancerologia | Instituto Nacional de Câncer | Ministério da Saúde