ARTIGO ORIGINAL
RET Proto-oncogene Sequencing in a Cohort of Patients with Medullary Thyroid Carcinoma in the State of Bahia, Brazil
Sequenciamento do Proto-oncogene RET em uma Coorte de Pacientes com Carcinoma Medular de Tireoide do Estado da Bahia, Brasil
Secuenciación del Protooncogén RET en una Cohorte de Pacientes con Carcinoma Medular de Tiroides del Estado de Bahía, Brasil
https://doi.org/10.32635/2176-9745.RBC.2024v70n4.4738
Rafael Reis Campos da Matta1, Marli Teresinha Viapiana Camelier2; Taíse Lima de Oliveira Cerqueira3; Jocyel Brito de Oliveira4; Juliana Lima Von Amon5; Ana Clara Tosta Telles6; Gilberto Dauricio Silva Leite7; Fabyan Esberard de Lima Beltrão8; Ana Luiza Silva Maia9; Helton Estrela Ramos10
1,4,5,6Universidade Federal da Bahia (UFBA), Instituto de Ciências da Saúde (ICS). Programa de Pós-Graduação em Processos Interativos dos Órgãos e Sistemas (PPGPIOS). Salvador (BA), Brasil. E-mails: rafaeldamatta19@gmail.com; jocyeloliveirabio@gmail.com; juammon@gmail.com; anaclaratelles@yahoo.com.br. Orcid iD: https://orcid.org/0009-0003-6378-8579; Orcid iD: https://orcid.org/0000-0002-1342-0368; Orcid iD: https://orcid.org/0000-0002-8513-992X; Orcid iD: https://orcid.org/0000-0002-2855-5855
2Hospital de Clínicas de Porto Alegre (HCPA), Serviço de Endocrinologia. Porto Alegre (RS) Brasil. E-mail: marlivcamelier@gmail.com. Orcid iD: https://orcid.org/0000-0002-9581-4765
3,7UFBA/ICS/Departamento de Biorregulação. Salvador (BA), Brasil. E-mails: taisellima@hotmail.com; gilberto.leite@ufba.br. Orcid iD: https://orcid.org/0009-0007-8658-1799; Orcid iD: https://orcid.org/0009-0004-6012-0669
8Universidade Federal da Paraíba (UFPB), Hospital Universitário Lauro Wanderley (HULW). João Pessoa (PB), Brasil. E-mail: fesberard@uol.com.br. Orcid iD: https://orcid.org/0000-0001-9713-2584
9Universidade Federal do Rio Grande do Sul (UFRGS), Escola de Medicina, Departamento de Medicina Interna. HCPA, Serviço de Endocrinologia. Porto Alegre (RS), Brasil. E-mail: almaia@ufrgs.br. Orcid iD: https://orcid.org/0000-0002-4186-5532
10UFBA/ICS/Departamento de Biorregulação. PPGPIOS. Programa de Pós-Graduação em Medicina e Saúde (PPGMS). Salvador (BA), Brasil. E-mail: ramoshelton@gmail.com Orcid ID: https://orcid.org/0000-0002-2900-2099
Corresponding author: Rafael Reis Campos da Matta. Avenida Rui Barbosa, 430 – Centro. Muritiba (BA), Brasil. CEP 44340-000. E- mail: rafaeldamatta19@gmail.com
ABSTRACT
Introduction: Medullary thyroid carcinoma is a rare cancer that originates in parafollicular C cells and can be sporadic or hereditary. Both sporadic and hereditary diseases are primarily caused by mutations in the RET proto-oncogene. Objective: To investigate RET gene pathogenic germline variants in a cohort of patients with medullary thyroid carcinoma in the State of Bahia. Method: Cross-sectional, descriptive study involving patients with histopathological diagnosis of medullary thyroid carcinoma, referred for molecular testing from 2020 to 2022. Clinical and pathological charts were collected from medical data. Genomic DNA was extracted from peripheral blood. Exons 10, 11, 13, 14, and 15 of RET were amplified using polymerase chain reaction technique and subsequently sequenced using the Sanger method. Results: The study included 29 patients (82.8% women). The mean age at diagnosis was 46.5 ± 13.1 years, and the mean tumor size was 2.1 ± 1.4 cm. According to the TNM classification, 38% of tumors were staged as T1a, 27.6% as T1b, 24.1% as T2, and 10.3% as T3. Regional lymph node metastasis (N1) was present in 44.8% of the cases. Distant metastasis (M1) to the mediastinum was observed in one case (3.4%). Variants of RET were identified in 55.2% of the patients. The C634R pathogenic variant was identified in one patient (3.4%). Conclusion: This study managed to describe the clinical and molecular profile of patients with medullary thyroid carcinoma in the State of Bahia.
Key words: Carcinoma, Medullary; Thyroid Neoplasms; Mutation; Multiple Endocrine Neoplasia Type 2a; Polymorphism, Genetic.
RESUMO
Introdução: O carcinoma medular da tireoide é um câncer raro que se origina nas células C parafoliculares e pode ser esporádico ou hereditário. Tanto as doenças esporádicas quanto as hereditárias são causadas principalmente por mutações no proto-oncogene RET. Objetivo: Investigar variantes germinativas patogênicas do gene RET em uma coorte de pacientes com carcinoma medular da tireoide no Estado da Bahia. Método: Estudo transversal, descritivo, envolvendo pacientes com diagnóstico histopatológico de carcinoma medular da tireoide, encaminhados para testes moleculares de 2020 a 2022. Dados clínicos e patológicos foram coletados de dados médicos. O DNA genômico foi extraído do sangue periférico. Os éxons 10, 11, 13, 14 e 15 do RET foram amplificados usando a técnica de reação em cadeia da polimerase e posteriormente sequenciados usando o método de Sanger. Resultados: O estudo incluiu 29 pacientes (82,8% mulheres). A idade média no diagnóstico foi de 46,5 ± 13,1 anos, e o tamanho médio do tumor foi de 2,1 ± 1,4 cm. De acordo com a classificação TNM, 38% dos tumores foram estadiados como T1a, 27,6% como T1b, 24,1% como T2 e 10,3% como T3. Metástase linfonodal regional (N1) esteve presente em 44,8% dos casos. Metástase distante (M1) para o mediastino foi observada em um caso (3,4%). Variantes do RET foram identificadas em 55,2% dos pacientes. A variante patogênica C634R foi identificada em um paciente (3,4%). Conclusão: Este estudo conseguiu descrever o perfil clínico e molecular de pacientes com carcinoma medular de tireoide no Estado da Bahia.
Palavras-chave: Carcinoma Medular; Neoplasias da Glândula Tireoide; Mutação; Neoplasia Endócrina Múltipla Tipo 2a; Polimorfismo Genético.
RESUMEN
Introducción: El carcinoma medular de tiroides es un cáncer poco común que se origina en las células C parafoliculares y puede ser esporádico o hereditario. Tanto las enfermedades esporádicas como las hereditarias son causadas principalmente por mutaciones en el protooncogén RET. Objetivo: Investigar variantes patogénicas de la línea germinal del gen RET en una cohorte de pacientes con carcinoma medular de tiroides en el estado de Bahía. Método: Estudio descriptivo transversal que involucró a pacientes con diagnóstico histopatológico de carcinoma medular de tiroides, remitidos para pruebas moleculares del 2020 al 2022. Las informaciones clínicas y patológicas se recolectaron a partir de datos médicos. El ADN genómico se extrajo de sangre periférica. Los exones 10, 11, 13, 14 y 15 del RET se amplificaron mediante la técnica de reacción en cadena de la polimerasa y posteriormente se secuenciaron mediante el método de Sanger. Resultados: Se incluyeron 29 pacientes (82,8% mujeres). La edad promedio al diagnóstico fue de 46,5 ± 13,1 años y el tamaño promedio del tumor fue de 2,1 ± 1,4 cm. Según la clasificación TNM, el 38% de los tumores se estadificaron como T1a, el 27,6% como T1b, el 24,1% como T2 y el 10,3% como T3. La metástasis a los ganglios linfáticos regionales (N1) estuvo presente en el 44,8% de los casos. En un caso (3,4%) se observó metástasis a distancia (M1) al mediastino. Se identificaron variantes del RET en el 55,2% de los pacientes. La variante patogénica C634R se identificó en un paciente (3,4%). Conclusión: Este estudio logró describir el perfil clínico y molecular de los pacientes con carcinoma medular de tiroides en el estado de Bahía.
Palabras clave: Carcinoma Medular; Neoplasias de la Tiroides; Mutación; Neoplasia Endocrina Múltiple Tipo 2ª;. Polimorfismo Genético.
INTRODUCTION
Medullary thyroid cancer (MTC) is a rare malignant tumor derived from parafollicular C cells. It accounts for 3-5% of all thyroid malignancies and is estimated to be diagnosed in 0.4 to 1.4% of patients with thyroid nodules1. The 10-year mortality rate for MTC ranges from 13 to 38% and is responsible for 15% of thyroid cancer-related deaths2.
MTC can manifest as a sporadic disease (accounting for 75-80% of the cases) or as a component of an inherited disorder known as multiple endocrine neoplasia type 2 (MEN2) (occurring in 20-25% of the cases)3. Both sporadic and hereditary forms of the disease are primarily caused by mutations in the RET (REarranged during Transfection) proto-oncogene.
The RET proto-oncogene was first identified in 1985 from the transfection of NIH 3T3 cells with human lymphoma DNA4. RET is located on human chromosome 10 (10q11.2), consists of 21 exons and is estimated to be approximately 52 kb in size5. It encodes the RET receptor tyrosine kinase, a transmembrane protein expressed primarily in neural crest and urogenital cells6-8.
About 98% of patients with hereditary MTC have germline mutations in exons 10, 11, 13, 14, 15 and 16 of the RET gene9. Rare germline mutations in exons 5 and 8 have been identified in some familial tumors10,11. In 95% of the patients with MEN2A, RET mutations are found in codons 609, 611, 618, and 620 of exon 10 or in codon 634 of exon 1112,13. Mutations in codon 634 are the most frequent, occurring in more than 80% of MEN2A cases14. The C634R mutation is the most common mutation at codon 634 and accounts for 50% of all mutations in MEN2A15. Among sporadic cases, approximately 23-66% of the patients have the somatic mutation M918T in RET exon 1616,17.
The identification of mutations in the RET gene combined with a complete clinical assessment of the patient forms the basis for individualized treatment in MTC. Therefore, genetic screening for RET is considered a key tool for the diagnosis of MTC and has important implications for the clinical management of patients and their families18.
This study aimed to report the clinical and molecular characteristics of patients with MTC in the State of Bahia.
METHOD
Cross-sectional and descriptive study involving 29 patients with MTC referred for RET molecular testing by endocrinologists, oncologists, and head and neck surgeons at various public and private healthcare institutions in the State of Bahia between 2020 to 2022. The inclusion criteria were patients: a) aged 18 years or older, and b) with confirmed MTC diagnosis based on histopathological reports. There were no exclusion criteria.
DNA was extracted from peripheral blood leukocytes using the PureLink Genomic DNA Mini Kit (Invitrogen, Carlsbad, USA), following the manufacturer's instructions. DNA fragments corresponding to exons 10, 11, 13, 14, and 15 of RET were separately amplified using the polymerase chain reaction (PCR) technique with specific primers. The PCR conditions were previously published19. The sequences of the primers used in the PCR are shown in Table 1.
Table 1. Sequences of each primer used in polymerase chain reaction
|
Exon |
Primer |
Sequence |
|
10 |
F R |
5’ -TTGCGACACCAGTTGCCGAG- 3’ 5’ -CAGCAATTTCCTCCCTTGTTG- 3’ |
|
11 |
F R |
5’ -GAGCCATGAGGCAGAGCATA- 3’ 5’ -CCCTCACCAGGATCTTGAAGG- 3’ |
|
13 |
F R |
5’ -TGAACTTGGGCAAGGCGATG- 3’ 5’ -GGGAGAACAGGGCTGTATGGAG- 3’ |
|
14 |
F R |
5’ -AAGACCCAAGCTGGCTGAG- 3’ 5’ -GCTGGGTGCAGAGCCATAT- 3’ |
|
15 |
F R |
5’ -CTCTGCTGGTCACACCAGGC- 3’ 5’ -GGTATCTTTCCTAGGCTTCCC-3’ |
Purified PCR products were directly sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit and the ABI 3500 Genetic Analyzer (Applied Biosystems, Foster City, USA). Each sequenced sample was analyzed using the BioEdit Sequence Alignment Editor software, version 7.2.520. Subsequently, the samples were submitted to germline mutation analyses for exons 10, 11, 13, 14, and 15 of RET (RefSeq:NM_020975.6) using the Basic Local Alignment Search Tool21.
The identified RET proto-oncogene variants were classified as benign, pathogenic or of uncertain significance according to the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP)22 guidelines and the ClinVar database23.
Descriptive statistics was utilized to evaluate the data with means, medians, and standard deviations calculated for the quantitative variables. Pearson's test was applied to evaluate the linear correlation coefficient among the variables analyzed. A significance level of p < 0.05 was accepted as statistically significant. The analyses were performed in the statistical program GraphPad Prism, v.7.0024.
The Research Ethics Committee of “Instituto de Ciências da Saúde” of “Universidade Federal da Bahia” approved the study, report 5771867 (CAAE (submission for ethical review) 46193121,2,0000,5662) in compliance with Directive 466/1225 of the National Health Council. All the procedures were conducted according to the Declaration of Helsinki. Written informed consent was obtained from all study participants.
RESULTS
The study sample comprised 29 patients diagnosed with MTC, consisting in 24 women (82.8%) and 5 men (17.2%). The mean age at diagnosis was 46.5 ± 13.1 years (median 46, range 20-70). The mean tumor size was 2.1 ± 1.4 cm (median 1.8, range 0.1-5.0). Most tumors were unifocal (79.3%) and non-encapsulated (89.7%).
Six variants of the RET were identified in 16 patients
(55.2%), consisting of one pathogenic (C634R), four
benign (G691S, L769L, S836S and S904S), and one variant of uncertain
significance (Y791N). Specifically, the pathogenic
variant C634R was identified in one patient (3.4%). The benign variants G691S,
L769L, S836S and S904S, described as RET single nucleotide polymorphisms (SNP),
were present in 15 patients (51.7%). Of these variants, the most frequent was
L769L, found as the sole RET variant in eight patients (27.6%). The
Y791N variant of uncertain significance was observed co-segregating with the
L769L variant in one patient (3.4%). Table 2 provides a
detailed breakdown of the clinical, pathological and molecular characteristics
of the 29 patients.
Table 2. Clinicopathological and molecular findings in 29 cases of MTC in the State of Bahia
|
Parameters |
Number of patients (%) |
|
Age at diagnosis (years) |
46.5 ± 13.1 |
|
Tumor size (cm) |
2.1 ± 1.4 |
|
Multifocality |
6 (20.7%) |
|
Tumor capsule |
3 (10.3%) |
|
Capsular invasion |
4 (13.8%) |
|
Extrathyroidal invasion |
1 (3.4%) |
|
Angiolymphatic invasion |
2 (6.9%) |
|
TNM Staging |
|
|
T1a |
11 (38%) |
|
T1b |
8 (27.6%) |
|
T2 |
7 (24.1%) |
|
T3 |
3 (10.3%) |
|
N0 |
16 (55.2%) |
|
N1 |
13 (44.8%) |
|
N1a |
4 of 13 (30.8%) |
|
N1b |
4 of 13 (30.8%) |
|
Mx |
28 (96.6%) |
|
M1 |
1 (3.4%) |
|
Association with PTC |
8 (27.6%) |
|
Hashimoto's thyroiditis |
8 (27.6%) |
|
RET germline variants |
|
|
C634R (exon 11) |
1 (3.4%) |
|
G691S (exon 11) + L769L (exon 13) + S904S (exon 15) |
1 (3.4%) |
|
G691S (exon 11) + S904S (exon 15) |
3 (10.3%) |
|
L769L (exon 13) |
8 (27.6%) |
|
L769L (exon 13) + S836S (exon 14) |
2 (6.9%) |
|
L769L (exon 13) + Y791N (exon 13) |
1 (3.4%) |
Caption: PTC = papillary thyroid carcinoma.
Correlation tests among clinical parameters and among clinical and molecular parameters were performed. First, the presence of lymph node metastasis was investigated to find whether there was association with age at diagnosis and tumor size. For this purpose, the patients were divided into two subgroups: without lymph node metastasis and with lymph node metastasis. As a result, no differences were observed in age at diagnosis (45.6 ± 10.8 vs. 47.6 ± 15.7 years, p = 0.691) and tumor size (1.53 ± 1.0 vs. 2.73 ± 1.5 cm, p = 0.016) between the subgroups.
Next, the presence of RET polymorphisms was evaluated for potential association with age and tumor size. For this purpose, patients were divided into two subgroups: without polymorphisms and with polymorphisms. As a result, no differences in age at diagnosis (48.7 ± 12.8 vs. 44.4 ± 13.3 years; p = 0.375) and tumor size (2.25 ± 1.59 vs. 2.0 ± 1.89 cm; p = 0.486) were found between the subgroups, respectively. The results of the correlation tests performed are shown in Figure 1.
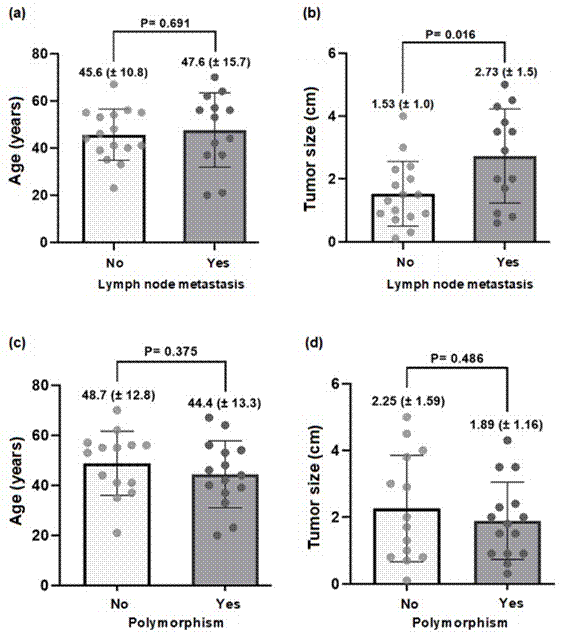
Figure 1. Association between presence/absence of lymph node metastasis and age at diagnosis (a) and tumor size (b) and between presence/absence of RET polymorphisms and age at diagnosis (c) and tumor size (d)
DISCUSSION
MTC is a tumor with peculiar characteristics and variable clinical presentation, depending on the causative RET mutation. In Brazil, especially in the Northeast region, records on the incidence, clinicopathological characteristics and genetic screening of MTC are still scarce. For the first time, the clinical-pathological and molecular characteristics of MTC patients in the State of Bahia were presented.
Although the sample size was small, it aligns with other studies that have demonstrated female predominance26-28. In hereditary MTC, there's no gender bias due to its autosomal dominant inheritance pattern. However, in sporadic MTC, the male-to-female ratio is about 1:1.4, indicating a slight female predominance29. Furthermore, thyroid cancer, in general, affects women more frequently and ranks as the fifth most common cancer in women in the Brazilian Northeast Region (7.98/100,000)30.
Although it can occur at any age, MTC has a peak incidence between the 4th and 5th decade of life¹, with a mean age at diagnosis between 50 and 54 years31. In this study, the mean age at diagnosis was compatible with the incidence range described in the literature and remained close to the values observed in other Brazilian studies27,32 and in studies conducted in China33, USA34 and Costa Rica35.
MTC is usually characterized by regional lymph node involvement early in its course. Approximately 35% to 50% of patients have lymph node involvement at initial diagnosis36. Lymph node involvement is considered an important prognostic factor, as it is associated with a lower biochemical cure rate and long-term survival37,38.
The presence of lymph node metastasis was investigated to find whether an association with age at diagnosis and tumor size was found. Although no correlation between these variables existed, studies have shown that lymph node metastases were found in 20-30% of the patients with tumors < 1 cm in diameter, in 50% of patients with tumors > 1 to 4 cm in diameter, and in up to 90% of patients with tumors > 4 cm39,40.
The C634R variant was found in a 41-year-old male at the time of diagnosis, and he also had a history of bilateral pheochromocytoma and chronic cholecystitis. Collectively, the clinical, pathological, and molecular characteristics in this patient were indicative of multiple endocrine neoplasia type 2A (MEN2A). MEN2 is a very rare disease. The reported incidence of this syndrome is one in every 80,000 live births and its global prevalence represents approximately 0.02-0.03% of all human tumors41, 42.
Mutations in codon 634 are highly prevalent in MEN2, detected in 30 to 50% of affected patients43. In Brazil, these mutations were observed in 262 patients (43.6%) as part of an extensive multicenter study44. Notably, the C634R variant ranked as the second most common among patients with MEN2A in two studies conducted in Rio Grande do Sul19,45 and was the third most frequent in Ceará27. The C634R variant was also the most frequent among patients with hereditary MTC in a European multicenter study46. C634R was also among the most common RET variants identified in MEN2A carrier families from Germany47, Italy48, and China49.
Although the absence of germline pathogenic variants in the remaining patients is suggestive of sporadic disease, some patients presented one or more features that suggest hereditary disease, as young age at diagnosis of MTC or multifocality of the tumor. It is possible that these apparently sporadic cases carry pathogenic variants in RET exons that were not analyzed in this study. Furthermore, mutations in other genes associated with MTC, presence of C cell hyperplasia and environmental characteristics of the region to which the sample belongs are some of the factors that may explain the characteristics observed in these patients.
The L769L SNP was the most prevalent in the current sample. This polymorphism was also the most frequent among patients with MEN2 at “Hospital das Clínicas de Porto Alegre“ in Rio Grande do Sul and at the “Hospital Universitário” of “Faculdade de Medicina de Ribeirão Preto” in São Paulo45. Similar to the studies by Siqueira et al.45 and Ceolin et al.50, both from Rio Grande do Sul, the co-segregation of the G691S/S904S SNPs was observed in the study sample.
The association between the presence of RET polymorphisms and susceptibility to the development and progression of MTC has been widely studied in recent decades. RET polymorphisms have been associated with early onset of MTC53, risk of developing pheochromocytoma54 and risk of metastatic disease55. In contrast, other studies have shown that there is no influence of RET polymorphisms on the clinical and oncological characteristics of MTC56-58. In the present study, no differences were found when the presence or absence of polymorphisms were related to age at diagnosis and tumor size. However, the small size of the present sample may have been a limiting factor for the proposed associations.
CONCLUSION
This study has outlined the clinical and molecular profile of MTC patients in the State of Bahia, Brazil. Additional studies with the same population are recommended to address other aspects of this rare condition in the State.
CONTRIBUTIONS
Rafael Reis Campos da Matta contributed to data collection, literature review and writing of the article. Marli Viapiana Camelier, Fabyan Esberard de Lima Beltrão and Ana Luiza Maia contributed to the literature review and writing of the article. Taíse Lima de Oliveira Cerqueira, Jocyel Brito de Oliveira, Juliana Lima Von Amon, Ana Clara Telles, Gilberto Dauricio Silva Leite contributed to the review and interpretation of the data. Helton Estrela Ramos contributed to the interpretation of the data, literature review and writing of the article. All the authors approved the final version for publication.
DECLARATION OF CONFLICT OF INTERESTS
There is no conflict of interests to declare.
FUNDING SOURCES
None.
REFERENCES
1. Viola D, Elisei R. Management of medullary thyroid cancer. Endocrinol Metab Clin North Am. 2019;48(1):285-301. doi: https://doi.org/10.1016/j.ecl.2018.11.006
2. Maia AL, Wajner SM, Vargas CV. Advances and controversies in the management of medullary thyroid carcinoma. Curr Opin Oncol. 2017;29(1):25-32. doi: https://doi.org/10.1097/CCO.0000000000000340
3. Ceolin L, Duval MADS, Benini AF, et al. Medullary thyroid carcinoma beyond surgery: advances, challenges, and perspectives. Endocr Relat Cancer. 2019;26(9):R499-518. doi: https://doi.org/10.1530/ERC-18-0574
4. Takahashi M, Ritz J, Cooper GM. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell. 1985;42(2):581-88. doi: https://doi.org/10.1016/0092-8674(85)90115-1
5. Kawai K, Takahashi M. Intracellular RET signaling pathways activated by GDNF. Cell Tissue Res. 2020;382(1):113-23. doi: https://doi.org/10.1007/s00441-020-03262-1
6. Takahashi M. Structure and expression of the ret transforming gene. IARC Sci Publ. 1988;(92):189-97.
7. Avantaggiato V, Dathan NA, Grieco M, et al. Developmental expression of the RET protooncogene. Cell Growth Differ. 1994;5(3):305-11.
8. Schuchardt A, D'Agati V, Larsson-Blomberg L, et al. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor RET. Nature. 1994;367(6461):380-83. doi: https://doi.org/10.1038/367380a0
9. Mohammadi M, Hedayati M. A brief review on the molecular basis of medullary thyroid carcinoma. Cell J. 2017;18(4):485-92. doi: https://doi.org/10.22074/cellj.2016.4715
10. Dvorakova S, Vaclavikova E, Duskova J, et al. Exon 5 of the RET proto-oncogene: a newly detected risk exon for familial medullary thyroid carcinoma, a novel germ-line mutation Gly321Arg. J Endocrinol Invest. 2005;28(10):905-9. doi: https://doi.org/10.1007/BF03345322
11. Silva AM, Maciel RM, Silva MR, et al. A novel germ-line point mutation in RET exon 8 (Gly(533)Cys) in a large kindred with familial medullary thyroid carcinoma. J Clin Endocrinol Metab. 2003;88(11):5438-43. doi: https://doi.org/10.1210/jc.2003-030997
12. Raue F, Frank-Raue K. Genotype-phenotype correlation in multiple endocrine neoplasia type 2. Clinics (Sao Paulo). 2012;67(Supl1):69-75. doi: https://doi.org/10.6061/clinics/2012(sup01)13
13. Wells SA Jr, Pacini F, Robinson BG, et al. Multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma: an update. J Clin Endocrinol Metab. 2013;98(8):3149-64. doi: https://doi.org/10.1210/jc.2013-1204
14. Chen H, Sippel RS, O'Dorisio MS, et al. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39(6):775-83. doi: https://doi.org/10.1097/mpa.0b013e3181ebb4f0
15. Maia AL, Siqueira DR, Kulcsar MA, et al. Diagnosis, treatment, and follow-up of medullary thyroid carcinoma: recommendations by the Thyroid Department of the Brazilian Society of Endocrinology and Metabolism. Arch Endocrinol Metab. 2014;58(7):667-700. doi: https://doi.org/10.1590/0004-2730000003427
16. Eng C, Mulligan LM, Smith DP, et al. Mutation of the RET protooncogene in sporadic medullary thyroid carcinoma. Genes Chromosomes Cancer. 1995;12(3):209-12. doi: https://doi.org/10.1002/gcc.2870120308
17. Marsh DJ, Learoyd DL, Andrew SD, et al. Somatic mutations in the RET proto-oncogene in sporadic medullary thyroid carcinoma. Clin Endocrinol (Oxf). 1996;44(3):249-57. doi: https://doi.org/10.1046/j.1365-2265.1996.681503.x
18. Romei C, Ciampi R, Elisei R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat Rev Endocrinol. 2016;12(4):192-202. doi: https://doi.org/10.1038/nrendo.2016.11
19. Puñales MK, Graf H, Gross JL, et al. RET codon 634 mutations in multiple endocrine neoplasia type 2: variable clinical features and clinical outcome. J Clin Endocrinol Metab. 2003;88(6):2644-9. doi: https://doi.org/10.1210/jc.2002-021422
20. BioEdit [Internet]. Versão 7.2.5. [sem local]: Informer Technologies, Inc.; ©2008-2024, [acesso 2024 jul 15]. Disponível em: https://bioedit.software.informer.com/7.2/
21. Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990;215(3):403-10. doi: https://doi.org/10.1016/s0022-2836(05)80360-2
22. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-24. doi: https://doi.org/10.1038/gim.2015.30
23. Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(Database issue):D980-5. doi: https://doi.org/10.1093/nar/gkt1113
24. Graph Pad: Prism [Internet]. Versão 7.0.0. Boston: GraphPad; 2017. [acesso 2024 jul 19]. Disponível em: https://www.graphpad.com/updates
25. Conselho Nacional de Saúde (BR). Resolução n° 466, de 12 de dezembro de 2012. Aprova as diretrizes e normas regulamentadoras de pesquisas envolvendo seres humanos. Diário Oficial da União, Brasília, DF. 2013 jun 13; Seção I:59.
26. Brandão LG, Cavalheiro BG, Junqueira CR. Prognostic influence of clinical and pathological factors in medullary thyroid carcinoma: a study of 53 cases. Clinics (Sao Paulo). 2009;64(9):849-56. doi: https://doi.org/10.1590/S1807-59322009000900005
27. Martins-Costa MC, Lindsey SC, Cunha LL, et al. A pioneering RET genetic screening study in the State of Ceará, Brazil, evaluating patients with medullary thyroid cancer and at-risk relatives: experience with 247 individuals. Arch Endocrinol Metab. 2018;62(6):623-35. doi: https://doi.org/10.20945/2359-3997000000088
28. Sermoud LM, Brochini MM, Rodrigues HD, et al. 15 years of medullary thyroid carcinoma (MTC) hospital morbidity according to Cancer Hospital-Based Registry in Brazil. Braz J Oncol. 2018;14(47):1-9. doi: https://doi.org/10.26790/BJO20181447A209
29. Raue F. German medullary thyroid carcinoma/multiple endocrine neoplasia registry. Langenbecks Arch Surg. 1998;383(5):334-6. doi: https://doi.org/10.1007/s004230050143
30. Instituto Nacional de Câncer. Estimativa 2023: Incidência de câncer no brasil [Internet]. Rio de Janeiro: Instituto Nacional de Câncer; 2022. [acesso 2023 dez 15]. Disponível em: https://www.inca.gov.br/publicacoes/livros/estimativa-2023-incidencia-de-cancer-no-brasil
31. Randle RW, Balentine CJ, Leverson GE, et al. Trends in the presentation, treatment, and survival of patients with medullary thyroid cancer over the past 30 years. Surgery. 2017;161(1):137-46. doi: https://doi.org/10.1016/j.surg.2016.04.053
32. Correia-Deur JE, Toledo RA, Imazawa AT, et al. Sporadic medullary thyroid carcinoma: clinical data from a university hospital. Clinics (Sao Paulo). 2009;64(5):379-86. doi: https://doi.org/10.1590/s1807-59322009000500002
33. Chow SM, Chan JK, Tiu SC, et al. Medullary thyroid carcinoma in Hong Kong Chinese patients. Hong Kong Med J. 2005;11(4):251-8.
34. Roman S, Lin R, Sosa JA. Prognosis of medullary thyroid carcinoma: demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer. 2006;107(9):2134-42. doi: https://doi.org/10.1002/cncr.22244
35. Calvo J, Torrealba G, Sáenz A, et al. Genetic and clinical features of medullary thyroid carcinoma: the experience of a single center in Costa Rica. J Cancer Epidemiol. 2016;2016:9637173. doi: https://doi.org/10.1155/2016/9637173
36. Sippel RS, Kunnimalaiyaan M, Chen H. Current management of medullary thyroid cancer. Oncologist. 2008;13(5):539-47. doi: https://doi.org/10.1634/theoncologist.2007-0239
37. Raue F, Kotzerke J, Reinwein D, et al. Prognostic factors in medullary thyroid carcinoma: evaluation of 741 patients from the German Medullary Thyroid Carcinoma Register. Clin Investig. 1993;71(1):7-12. doi: https://doi.org/10.1007/bf00210956b
38. Dralle H, Scheumann GF, Proye C, et al. The value of lymph node dissection in hereditary medullary thyroid carcinoma: a retrospective, European, multicentre study. J Intern Med. 1995;238(4):357-61. doi: https://doi.org/10.1111/j.1365-2796.1995.tb01210.x
39. Moley JF, DeBenedetti MK. Patterns of nodal metastases in palpable medullary thyroid carcinoma: recommendations for extent of node dissection. Ann Surg. 1999;229(6):880-7. doi: https://doi.org/10.1097/00000658-199906000-00016
40. Scollo C, Baudin E, Travagli JP, et al. Rationale for central and bilateral lymph node dissection in sporadic and hereditary medullary thyroid cancer. J Clin Endocrinol Metab. 2003;88(5):2070-5. doi: https://doi.org/10.1210/jc.2002-021713
41. Newey PJ. Multiple endocrine neoplasia. Medicine. 2021;49(9):539-43. doi: https://doi.org/10.1016/j.mpmed.2021.06.003
42. Romei C, Pardi E, Cetani F, et al. Genetic and clinical features of multiple endocrine neoplasia types 1 and 2. J Oncol. 2012;2012:1-15. doi: https://doi.org/10.1155/2012/705036
43. Maciel RMB, Maia AL. Global Endocrinology: geographical variation in the profile of ret variants in patients with medullary thyroid cancer: a comprehensive review. Eur J Endocrinol. 2021;186(1):R15-30. doi: https://doi.org/10.1530/EJE-21-0753
44. Maciel RMB, Camacho CP, Assumpção LVM, et al. Genotype and phenotype landscape of MEN2 in 554 medullary thyroid cancer patients: the BrasMEN study. Endocr Connect. 2019;8(3):289-98. doi: https://doi.org/10.1530/EC-18-0506
45. Siqueira DR, Ceolin L, Ferreira CV, et al. Role of RET genetic variants in MEN2-associated pheochromocytoma. Eur J Endocrinol. 2014;170(6):821-8. doi: https://doi.org/10.1530/EJE-14-0084
46. Machens A, Niccoli-Sire P, Hoegel J, et al. Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med. 2003;349(16):1517-25. doi: https://doi.org/10.1056/nejmoa012915
47. Frank-Raue K, Höppner W, Frilling A, et al. Mutations of the ret protooncogene in German multiple endocrine neoplasia families: relation between genotype and phenotype. German Medullary Thyroid Carcinoma Study Group. J Clin Endocrinol Metab. 1996;81(5):1780-3. doi: https://doi.org/10.1210/jcem.81.5.8626834
48. Romei C, Mariotti S, Fugazzola L, et al. ItaMEN network. Multiple endocrine neoplasia type 2 syndromes (MEN 2): results from the ItaMEN network analysis on the prevalence of different genotypes and phenotypes. Eur J Endocrinol. 2010;163(2):301-8. doi: https://doi.org/10.1530/eje-10-0333
49. Qi XP, Chen XL, Ma JM, et al. RET proto-oncogene genetic screening of families with multiple endocrine neoplasia type 2 optimizes diagnostic and clinical management in China. Thyroid. 2012;22(12):1257-65. doi: https://doi.org/10.1089/thy.2012.0134
50. Ceolin L, Siqueira DR, Ferreira CV, et al. Additive effect of RET polymorphisms on sporadic medullary thyroid carcinoma susceptibility and tumor aggressiveness. Eur J Endocrinol. 2012;166(5):847-54. doi: https://doi.org/10.1530/EJE-11-1060
51. Magalhães PK, Castro M, Elias LL,. Polymorphisms in the RET proto-oncogene and the phenotypic presentation of familial medullary thyroid carcinoma. Thyroid. 2004;14(10):848-52. doi: https://doi.org/10.1089/thy.2004.14.848
52. Wiench M, Wygoda Z, Gubala E, et al. Estimation of risk of inherited medullary thyroid carcinoma in apparent sporadic patients. J Clin Oncol. 2001;19(5):1374-80. doi: https://doi.org/10.1200/jco.2001.19.5.1374
53. Sromek M, Czetwertyńska M, Skasko E, etal. The frequency of selected polymorphic variants of the RET gene in patients with medullary thyroid carcinoma and in the general population of central. Poland. Endocr Pathol. 2010;21(3):178-85. doi: https://doi.org/10.1007/s12022-010-9125-8
54. Lebeault M, Pinson S, Guillaud-Bataille M, et al. Nationwide french study of RET variants detected from 2003 to 2013 suggests a possible influence of polymorphisms as modifiers. Thyroid. 2017;27(12):1511-22. doi: https://doi.org/10.1089/thy.2016.0399
55. Siqueira DR, Romitti M, Rocha AP, et al. The RET polymorphic allele S836S is associated with early metastatic disease in patients with hereditary or sporadic medullary thyroid carcinoma. Endocr Relat Cancer. 2010;17(4):953-63. doi: https://doi.org/10.1677/erc-09-0312
56. Machens A, Frank-Raue K, Lorenz K, et al. Clinical relevance of RET variants G691S, L769L, S836S and S904S to sporadic medullary thyroid cancer. Clin Endocrinol (Oxf). 2012;76(5):691-7. doi: https://doi.org/10.1111/j.1365-2265.2011.04293.x
57. Zhang Y, Wang S, Chen X, et al. Quantitative assessment of the association between L769L and S836S polymorphisms at RET gene and medullary thyroid carcinoma risk. Tumour Biol. 2014;35(7):6641-7. doi: https://doi.org/10.1007/s13277-014-1878-0
58. Skalniak A, Trofimiuk-Müldner M, Przybylik-Mazurek E, et al. Modifier role of common RET variants in sporadic medullary thyroid carcinoma. Int J Mol Sci. 2021;22(21):11794. doi: https://doi.org/10.3390/ijms222111794
Recebido em 10/6/2024
Aprovado em 3/9/2024
Associate-editor: Tatiana de Almeida Simão. Orcid iD: https://orcid.org/0000-0001-8509-2247
Scientific-editor: Anke Bergmann. Orcid iD: https://orcid.org/0000-0002-1972-8777
![]()
Este é um artigo publicado em acesso aberto (Open Access) sob a licença Creative Commons Attribution, que permite uso, distribuição e reprodução em qualquer meio, sem restrições, desde que o trabalho original seja corretamente citado.