RELATO DE CASO
Rabdomiossarcoma do Subtipo Alveolar em Região Tenar com Invasão Linfonodal Axilar em Adulto: Relato de Caso
Alveolar Subtype Rhabdomyosarcoma of the Thenar Region with Axillary Lymph Node Invasion in an Adult: Case Report
Rabdomiosarcoma del Subtipo Alveolar en la Región Tenar con Invasión de los Ganglios Linfáticos Axilares en Adulto: Informe de Caso
https://doi.org/10.32635/2176-9745.RBC.2025v71n2.4969
Aline Graciele Henriques Campos¹; Estela Cesar Oyama²; Maria Eduarda Pardal Simonato³; Maikely Bruna Leite4; Rafael Gomes Paz5; Victor Pereira da Silva6; Liara Camila Tusset7; Matheus Vieira da Costa8
1-6,8Universidade Estadual do Oeste do Paraná (Unioeste-PR). Francisco Beltrão (PR), Brasil. E-mails: alineghcampos3@gmail.com; estela.oyama19@gmail.com; mariaeduarda.psh@gmail.com; maikelyleite20@gmail.com; rafaelgomespaz@hotmail.com; silvapereiravictor@hotmail.com; matpatologia@yahoo.com.br. Orcid iD: https://orcid.org/0000-0002-3784-7569; Orcid iD: https://orcid.org/0009-0001-8387-1850; Orcid iD: https://orcid.org/0009-0003-7244-0750; Orcid iD: https://orcid.org/0000-0003-0023-5144; Orcid iD: https://orcid.org/0009-0003-3666-2399; Orcid iD: https://orcid.org/0009-0005-9907-584X; Orcid iD: https://orcid.org/0009-0009-4055-0705
7Centro Universitário de Pato Branco. Pato Branco (PR), Brasil. E-mail: liara.oncologia@gmail.com. Orcid iD: https://orcid.org/0009-0002-7904-6046
Endereço para correspondência: Aline Graciele Henriques Campos. Rua Bahia, 835, apto. 14 – Presidente Kennedy. Francisco Beltrão (PR), Brasil. CEP 85605-270. E-mail: alineghcampos3@gmail.com
RESUMO
Introdução: O rabdomiossarcoma é um subtipo raro de neoplasia maligna de tecidos moles cujas células são semelhantes a mioblastos. Representa 1% das neoplasias em adultos, com mau prognóstico e alta taxa de recidiva. Neste trabalho, objetiva-se relatar um caso ultrarraro de rabdomiossarcoma alveolar que acometeu a mão de uma paciente adulta, além de realizar uma revisão integrativa de casos semelhantes, haja vista que cânceres primários na mão têm incidência inferior a 1%. Relato do caso: Mulher, 33 anos, procurou atendimento clínico por presença de lesão arroxeada e não ulcerada em região de tenar da mão esquerda, com linfadenomegalia em axila esquerda. Inicialmente, a ressonância revelou uma formação expansiva na região tenar, na qual a biópsia incisional e o exame-histoquímico sugeriram rabdomiossarcoma alveolar de alto grau. Realizou-se amputação do primeiro metacarpo de mão esquerda e dissecção axilar dos linfonodos comprometidos, além de quimioterapia SAIME intercalada com SAVAC a cada três semanas, e radioterapia de intervalo, por seis meses. Tratamento sem complicações e atualmente sem recidivas. Conclusão: O caso relatado é de apresentação anatômica e faixa etária pouco comum para o rabdomiossarcoma, e os principais aspectos para bom seguimento clínico são o diagnóstico precoce e a terapia multimodal.
Palavras-chave: Rabdomiossarcoma; Neoplasias de Tecidos Moles; Adulto; Amputação Cirúrgica; Relato de Caso.
ABSTRACT
Introduction: Rhabdomyosarcoma is a rare subtype of soft tissue malignancy whose cells are similar to myoblasts. It represents 1% of neoplasms in adults, with a poor prognosis and high recurrence rate. In this work, the objective is to report an ultra-rare case of alveolar rhabdomyosarcoma that affected the hand of an adult patient, in addition to conducting an integrative review of similar cases, given that primary cancers in the hand have an incidence of less than 1%. Case report: A 33-year-old woman sought clinical care due to the presence of a purplish, non-ulcerated lesion in the thenar region of her left hand, with lymphadenomegaly in the left axilla. Initially, the MRI revealed an expansive formation in the thenar region, in which the incisional biopsy and histochemical examination suggested high-grade alveolar rhabdomyosarcoma. Amputation of the first metacarpal of the left hand and axillary dissection of the affected lymph nodes were performed, in addition to SAIME chemotherapy interspersed with SAVAC every three weeks, and interval radiotherapy for six months. Treatment without complications and currently without recurrences. Conclusion: The reported case has an unusual anatomical presentation and age range for rhabdomyosarcoma, and the main aspects for favorable clinical follow-up are early diagnosis and multimodal therapy.
Key words: Rhabdomyosarcoma; Soft Tissue Neoplasms; Adult; Surgical Amputation; Case Report.
RESUMEN
Introducción: El rabdomiosarcoma es un subtipo raro de neoplasia maligna de tejidos blandos cuyas células son similares a los mioblastos. Representa el 1% de las neoplasias en adultos, con mal pronóstico y alta tasa de recurrencia. En este trabajo el objetivo es reportar un caso extremadamente raro de rabdomiosarcoma alveolar que afectó la mano de un paciente adulto, además de realizar una revisión integradora de casos similares, dado que los cánceres primarios en la mano tienen una incidencia menor del 1%. Informe del caso: Mujer de 33 años acudió a atención clínica por la presencia de una lesión violácea no ulcerada en la región tenar de la mano izquierda, con linfadenomegalia en la axila izquierda. Inicialmente, la resonancia magnética reveló una formación expansiva en la región tenar, en el cual la biopsia incisional y el examen histoquímico sugirieron rabdomiosarcoma alveolar de alto grado. Se realizó amputación del primer metacarpio de la mano izquierda y disección axilar de los ganglios afectados, además de quimioterapia SAIME intercalada con SAVAC cada tres semanas y radioterapia interválica durante seis meses. Tratamiento sin complicaciones y actualmente sin recidivas. Conclusión: El caso reportado tiene una presentación anatómica y rango de edad inusual para rabdomiosarcoma, y los principales aspectos para un buen seguimiento clínico son el diagnóstico precoz y la terapia multimodal.
Palabras clave: Rabdomiosarcoma; Neoplasias de los Tejidos Blandos; Adulto; Amputación Quirúrgica; Informe de Caso.
INTRODUÇÃO
O rabdomiossarcoma (RMS) é um tumor maligno mesenquimatoso raro com diferenciação muscular esquelética1. A doença é localmente invasiva, o que demanda abordagem terapêutica multimodal. O prognóstico varia com o sítio de origem, tamanho do tumor, estadiamento clínico, idade do paciente e tipo histológico2,3. Com base nas características clinicopatológicas e nas anormalidades genéticas, há os subtipos embrionário (variação de células primitivas arredondadas até densamente eosinofílicas), alveolar (camadas de células de tamanho médio, células gigantes dispersas e formação de padrão alveolar), fusiforme ou esclerosante (células fusiformes em fascículos, com padrão esclerosante pseudovascular) e pleomórfico (rabdomioblastos atípicos)1,2,4. Representa 5% de todos os cânceres pediátricos, cujos subtipos mais comuns são o embrionário e alveolar5, e 1% das neoplasias em adultos, sendo a variante pleomórfica mais comum5. A incidência do rabdomiossarcoma alveolar (RMSa) é constante dos 0 aos 19 anos, com aproximadamente um caso por um milhão de crianças e adolescentes, e apresenta menor incidência após essa idade. Além disso, os tumores malignos primários que afetam a mão são extremamente raros, com incidência inferior a 1%6.
Assim, a partir dos dados expostos, objetiva-se relatar um caso ultrarraro de RMSa que acometeu a mão de uma paciente adulta, e serão discutidos o tratamento, os fatores de pior prognóstico e o acompanhamento ambulatorial, a fim de estabelecer um paralelo entre o que foi aplicado na paciente e que é descrito na literatura. Também objetiva-se fazer uma revisão integrativa de relatos de casos de RMS primário em extremidades de adultos. A pesquisa foi aprovada pelo Comitê de Ética em Pesquisa da Universidade Estadual do Oeste do Paraná sob o número de parecer 7050791 (CAAE: 82359624.7.0000.0107), de acordo com a Resolução do Conselho Nacional de Saúde (CNS) n.º 466/127.
RELATO DO CASO
Paciente feminina, 33 anos, procedente de Pato Branco (PR), casada, dois filhos, com histórico de hipotireoidismo e diabetes mellitus gestacional, em uso contínuo de levotiroxina sódica 75 mcg. Cirurgias prévias: cesariana e exérese de tumor de conjuntiva. Nega tabagismo e etilismo, e relata, no histórico familiar patológico, a avó com leucemia.
Em maio de 2021, procurou atendimento clínico ambulatorial por presença de lesão em região tenar da mão esquerda, arroxeada, não ulcerada, medindo cerca de 3,5 cm, associada à linfadenomegalia em axila esquerda. Foi realizada ressonância magnética da mão esquerda que demonstrou formação expansiva na região tenar medindo 3,3 x 3,0 x 2,2 cm. Foi feita uma biópsia incisional que demonstrou neoplasia pouco diferenciada, e o exame imuno-histoquímico sugeriu RMS de alto grau, do subtipo alveolar. Foi realizado estadiamento, com tomografias de tórax e abdome total, o qual apresentou apenas linfadenomegalia axilar esquerda já identificada em exame físico, medindo 3,3 x 2,0 cm, resultando, assim, em estadiamento clínico III cT1aN1M0 de risco intermediário.
Realizou-se a cirurgia em maio de 2023 up-front com amputação do primeiro metacarpo de mão esquerda e linfadenectomia axilar esquerda, cujo anatomopatológico, na Figura 1, revelou RMSa pouco diferenciado, medindo 3,7 cm, não infiltrativo em tecido ósseo e com margens livres, com 18 linfonodos positivos do total de 43. Pós-operatório sem complicações, com tempo de internamento total de dois dias. A paciente realizou protocolo de quimioterapia SAIME (etoposideo + ifosfamida + mesna), intercalado com SAVAC (vincristina + doxorrubicina + ciclofosfamida) a cada três semanas, com radioterapia de intervalo, por seis meses. Atualmente sem recidivas.
DISCUSSÃO
O RMS de extremidades constitui uma minoria de casos relatados na literatura – menos de um terço dos sarcomas – e normalmente se apresenta como doença disseminada ao diagnóstico, com prognóstico pior em comparação com RMS de outros locais8,9. Os fatores de risco para a doença são genéticos, como a presença da síndrome de Li-Fraumeni, neurofibromatose tipo 1, síndrome de Costello e síndrome de Noonan; e ambiental, como exposição pré-natal aos raios-X, uso parental de drogas, alergias infantis, uso de medicamentos para fertilidade e nascimento prematuro8. A clínica é associada ao surgimento de massas de tecidos moles, frequentemente indolores, e podem surgir sintomas compressivos. O diagnóstico é por biópsia incisional, excisional ou com agulha grossa, e posterior análise histológica e molecular. O RMSa é composto por células redondas e densamente compactadas que revestem septações semelhantes aos alvéolos pulmonares8. Essas características estão presentes na biópsia da paciente deste relato que, conforme a Figura 1, há células grandes, redondas a poligonais com bordas definidas, e pouca variação no tamanho individual das células tumorais, com citoplasma granular eosinofílico abundante, núcleo redondo e vesicular com nucléolo proeminente com crescimento em ninhos ou organoides.
O tratamento para adultos é semelhante à terapia pediátrica, com abordagem multimodal por meio de ressecção cirúrgica, quimioterapia para citorredução primária e erradicação de doença metastática, e radioterapia ionizante para o controle de doença residual microscópica, conforme o estadiamento, a variedade patológica e a molecular. Essa terapêutica multimodal apresenta maior eficácia clínica, com menor toxicidade do tratamento10. No caso da paciente, não foi realizada análise molecular do tumor, mas o uso de dois protocolos de quimioterapia adjuvante e radioterapia potencialmente supera os mecanismos de resistência do tumor ao tratamento, visto que agem em diferentes vias de sinalização celular a fim de aumentar eficácia clínica com diminuição dos efeitos colaterais11. Esse tratamento agressivo e coordenado demonstrou ser eficaz, já que não há sinais de recidiva após mais de seis meses do tratamento.
Os casos de RMSa em adultos apresentam pior prognóstico se histologia alveolar ou localização anatômica desvantajosa, como extremidades, conforme neste caso relatado. Outros fatores de mau prognóstico são: idade avançada, sexo feminino, presença de metástases a distância no diagnóstico, tamanho do tumor primário superior a 5 cm na doença localizada; todos associados a adultos10. No caso relatado, o tumor primário de 3,7 cm e a localização em mão esquerda, embora menos comuns, foram associados a envolvimento linfonodal significativo, o que reforça a importância da terapia multimodal agressiva.
A Tabela 112-24 apresenta relatos e séries de casos de RMS primários em extremidades, em indivíduos maiores de 18 anos, recuperados no PubMed.gov, disponíveis gratuitamente na íntegra. Foram utilizados os seguintes descritores e marcadores booleanos: rhabdomyosarcoma AND hand OR foot OR extremity para a estratégia de busca.
CONCLUSÃO
O caso relatado é de apresentação anatômica e faixa etária pouco comum para o RMSa, e os principais aspectos para bom seguimento clínico são o diagnóstico precoce e a terapia multimodal.
CONTRIBUIÇÕES
Todos os autores contribuíram substancialmente na concepção e no planejamento do estudo; na obtenção, análise e interpretação dos dados; na redação e revisão crítica; e aprovaram a versão final a ser publicada.
DECLARAÇÃO DE CONFLITO DE INTERESSES
Nada a declarar.
FONTES DE FINANCIAMENTO
Não há.
REFERÊNCIAS
1. Kumar V, Aster JC, Abbas AK. Robbins Patologia Básica. 10. ed. Rio de Janeiro: Guanabara Koogan; 2021. 1421 p.
2. Cassidy J, Bissett D, Spence RAJ, et al. Oxford handbook of oncology. 4. ed. Oxford: Oxford University Press; 2015.
3. Agarwala S. Pediatric rhabdomyosarcomas and nonrhabdomyosarcoma soft tissue sarcoma. J Indian Assoc Pediatr Surg [Internet]. 2006 [acesso 2024 maio 3];11(1):15. Disponível em: http://www.jiaps.com/text.asp?2006/11/1/15/24632
4. Agaram NP. Evolving classification of rhabdomyosarcoma. Histopathology [Internet]. 2022 [acesso 2024 maio 3];80(1):98-108. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9425116/
5. Egas-Bejar D, Huh WW. Rhabdomyosarcoma in adolescent and young adult patients: current perspectives. Adolesc Health Med Ther [Internet]. 2014 [acesso 2024 maio 3];5:115-25. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4069040/
6. Xarchas K, Papavassiliou N, Tsoutseos N, et al. Rhabdomyosarcoma of the hand. Two case reports and a review of the literature. J Hand Surg Edinb Scotl. 1996;21(3):325-9.
7. Conselho Nacional de Saúde (BR). Resolução n° 466, de 12 de dezembro de 2012. Aprova as diretrizes e normas regulamentadoras de pesquisas envolvendo seres humanos. Diário Oficial da União, Brasília, DF. 2013 jun 13; Seção I:59.
8. Skapek SX, Ferrari A, Gupta AA, et al. Rhabdomyosarcoma. Nat Rev Dis Primer. 2019;5(1):1.
9. Seyhan N, Keskin M, Tosun Z, et al. Rhabdomyosarcoma in an adult hand. J Plast Surg Hand Surg. 2013;47(5):422-4.
10. Okcu MF. Rhabdomyosarcoma in childhood, adolescence, and adulthood: treatment. Uptodate [Internet]. 2023 [acesso 2024 maio 3];1. Disponível em: https://www.uptodate.com/contents/rhabdomyosarcoma-in-childhood-adolescence-and-adulthood-treatment
11. Van Erp A, Versleijen-Jonkers Y, Van der Graaf W, et al. Targeted therapy–based combination treatment in rhabdomyosarcoma. Mol Cancer Ther. 2018;17:1365-80.
12. Taza F, Kanwal A, Zulty M, et al. High-grade pleomorphic rhabdomyosarcoma in a 60-year-old male: a case report and review of the literature. J Community Hosp Intern Med Perspect. 2020;10(3):287-9. doi: https://www.doi.org.br/10.1080/20009666.2020.1766820
13. Gorunova L, Bjerkehagen B, Micci F, et al. Cytogenetic and molecular study of an adult sclerosing rhabdomyosarcoma of the extremity: MYOD1-mutation and clonal evolution. Cancer Genom Proteom. 2020;17(5):563-9. doi: https://www.doi.org/10.21873/cgp.20212
14. Widikusumo A, Triyanto L, Istutiningrum R, et al. Adult alveolar rhabdomyosarcoma on extremity, successful treatment with radiotherapy following chemotherapy: serial case report. Int J Appl Basic Med Res. 2019;9(2):121-3. doi: https://www.doi.org/10.4103/ijabmr.IJABMR_100_18
15. Martorell M, Ortiz CM, Garcia JA. Testicular fusocellular rhabdomyosarcoma as a metastasis of elbow sclerosing rhabdomyosarcoma: a clinicopathologic, immunohistochemical and molecular study of one case. Diagn Pathol. 2010;5(52):1-6. doi: https://www.doi.org/10.1186/1746-1596-5-52
16. Othman H, Mohamed Haflah NH, Sani MH, et al. Distal tibiofibular joint reconstruction using autograft in a rare case of lower limb sclerosing/spindle cell rhabdomyosarcoma: a two-year follow-up. Cureus. 2023;15(8):e42869. doi: https://www.doi.org/10.7759/cureus.42869
17. Rasekhi RT, Hosseini N, Babapoor-Farrokhran S, et al. A rare case of primary adult cardiac rhabdomyosarcoma with lower extremity metastasis. Radiol Case Rep. 2023;18(5):1666-70.
18. Widikusumo A, Triyanto L, Istutiningrum R, et al. Adult alveolar rhabdomyosarcoma on extremity, successful treatment with radiotherapy following chemotherapy: serial case report. Int J Appl Basic Med Res. 2019;9(2):121-3.
19. Bar Y, Merimsky O. Soft-Tissue sarcoma following traumatic injury: case report and review of the literature. Front Oncol. 2017;7:134. doi: https://www.doi.org/10.3389/fonc.2017.00134
20. Fang AS, Morse LJ, Wustrack R, et al. Complete regression of rhabdomyosarcoma in an adult secondary to postoperative wound infection following limb salvage surgery: a case report. Perm J. 2020;25(1):1. doi: https://www.doi.org/10.7812/TPP/20.172
21.Valone III F, Liu J, Genrich G, et al. Alveolar rhabdomyosarcoma causing acute compartment syndrome of the forearm: a case report and review of the literature. J Hand Microsurg. 2014;6(2):92-5. doi: https://www.doi.org/10.1007/s12593-013-0108-0
22. Gorunova L, Bjerkehagen B, Micci F, et al. Cytogenetic and molecular study of an adult sclerosing rhabdomyosarcoma of the extremity: MYOD1-mutation and clonal evolution. Cancer Genom Proteom. 2020;17(5):563-9. doi: https://www.doi.org/10.21873/cgp.20212
23. Fabian ID, Hildebrand GD, Wilson S, et al. Alveolar rhabdomyosarcoma of the foot metastasizing to the Iris: report of a rare case. BMC Cancer. 2016;16(447):1-3. doi: https://www.doi.org/10.1186/s12885-016-2496-6
24. Fisher MH, Woods JFC, Bartlett EK, et al. A rare case of rhabdomyosarcoma identified in a VRAM flap after lower extremity reconstruction. J Surg Case Rep. 2023;2023(3):rjad083. doi: https://www.doi.org/10.1093/jscr/rjad083
Aprovado em 19/2/2025
Editora-científica: Anke Bergmann. Orcid iD: https://orcid.org/0000-0002-1972-8777
Tabela 1. Relatos de casos de rabdomiossarcoma primário em extremidades corporais de maiores de 18 anos, publicados até janeiro de 2025
|
Autor |
Sexo/ idade (anos) |
Número de casos |
Local do rabdomiossarcoma primário |
Tamanho do sarcoma (cm) |
Subtipo histológico |
Metástases |
Tratamento |
Seguimento (meses) |
Resultado clínico |
|
|
12 |
M/60 |
1 |
Coxa direita |
4,5 |
Pleomórfico |
Pulmão |
C + QT + RT |
30 |
Remissão |
|
|
13 |
M/30 |
1 |
Pé esquerdo |
11 |
Esclerosante |
- |
C |
28 |
Remissão |
|
|
14 |
F/33* |
2 |
Coxa direita/Tríceps direito |
6x4x3/6x6,5x4 |
Alveolar |
- |
C + QT + RT / QT + RT |
- |
Remissão |
|
|
15 |
M/37 |
1 |
Cotovelo direito |
9x6 |
Esclerosante |
Testículo, mediastino, pulmão e coxa |
C + QT |
19 |
Óbito |
|
|
16 |
F/44 |
1 |
Tornozelo direito |
11 |
Esclerosante |
- |
QT + C + RT |
24 |
Remissão |
|
|
17 |
M/36 |
1 |
Perna esquerda |
4×3×2,8 |
Esclerosante |
- |
C + QT + RT |
9 |
Remissão |
|
|
18
|
F/33* |
2 |
Coxa direita/ Tríceps direito |
6×4×3 / 6×6,5×4 |
Alveolar |
- |
C + QT + RT |
9* |
Remissão |
|
|
19 |
M/31 |
1 |
Panturrilha direita |
- |
Pleomórfico |
- |
QT + C |
- |
- |
|
|
20 |
M/33 |
1 |
Compartimento anterior da perna |
12x10x25 |
Embrionário |
- |
QT + RT + C |
48 |
Remissão |
|
|
21 |
M/18 |
1 |
Antebraço esquerdo |
6,7x6,9x5,9 |
- |
- |
QT + C |
- |
- |
|
|
22 |
M/30 |
1 |
Pé esquerdo |
11 |
Esclerosante |
- |
- |
- |
- |
|
|
23 |
F/18 |
1 |
Pé |
- |
Alveolar |
Íris, membro inferior, cadeia inguinal externa, mediastino, pulmão |
C + QT |
26 |
Óbito |
|
|
24 |
M/36 |
1 |
Perna esquerda |
4x3x2,8 |
Esclerosante |
- |
QT + C + RT |
- |
Remissão |
|
Legendas: * = média; F = sexo feminino; M = sexo masculino; RT = radioterapia; C = cirurgia; QT = quimioterapia.
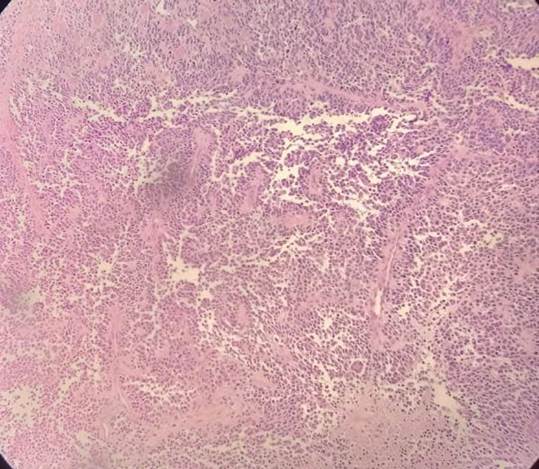
Figura 1. Anatomopatológico demonstrando rabdomiossarcoma alveolar pouco diferenciado
![]()
Este é um artigo publicado em acesso aberto (Open Access) sob a licença Creative Commons Attribution, que permite uso, distribuição e reprodução em qualquer meio, sem restrições, desde que o trabalho original seja corretamente citado.